 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1n6f | ||||||
|---|---|---|---|---|---|---|---|
| Title | tricorn protease in complex with Z-Phe-diketo-Arg-Glu-Phe | ||||||
 Components Components | tricorn protease | ||||||
 Keywords Keywords | HYDROLASE / tricorn protease / propeller | ||||||
| Function / homology |  Function and homology information Function and homology informationHydrolases; Acting on peptide bonds (peptidases); Serine endopeptidases / serine-type peptidase activity / proteolysis / cytoplasm Similarity search - Function | ||||||
| Biological species |   Thermoplasma acidophilum (acidophilic) Thermoplasma acidophilum (acidophilic) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / direct refinement / Resolution: 2.7 Å X-RAY DIFFRACTION / SYNCHROTRON / direct refinement / Resolution: 2.7 Å | ||||||
 Authors Authors | Kim, J.-S. / Groll, M. / Huber, R. / Brandstetter, H. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2002 Title: Navigation Inside a Protease: Substrate Selection and Product Exit in the Tricorn Protease from Thermoplasma acidophilum Authors: Kim, J.-S. / Groll, M. / Musiol, H.-J. / Behrendt, R. / Kaiser, M. / Moroder, L. / Huber, R. / Brandstetter, H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1n6f.cif.gz | 1.2 MB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1n6f.ent.gz | 989 KB | Display | PDB format |
| PDBx/mmJSON format | 1n6f.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/n6/1n6fftp://data.pdbj.org/pub/pdb/validation_reports/n6/1n6f | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1n6dC  1n6eC  1k32S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |







-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 121779.531 Da / Num. of mol.: 6 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Thermoplasma acidophilum (acidophilic) / Plasmid: pRSet6c / Species (production host): Escherichia coli / Production host:  References: UniProt: P96086, Hydrolases; Acting on peptide bonds (peptidases); Serine endopeptidases #2: Chemical | ChemComp-DKT / 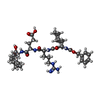  Mass: 771.900 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C38H57N7O10 Mass: 771.900 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C38H57N7O10#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 699 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 699 / Source method: isolated from a natural source / Formula: H2OHas protein modification | N | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.56 Å3/Da / Density % sol: 51.5 % | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 290 K / Method: vapor diffusion, hanging drop / pH: 6.4 Details: Isopropanol, MES, pH 6.4, VAPOR DIFFUSION, HANGING DROP, temperature 290K | |||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 7.5 | |||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: MPG/DESY, HAMBURG  / Beamline: BW6 / Wavelength: 1.05 Å / Beamline: BW6 / Wavelength: 1.05 Å |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Mar 9, 2002 / Details: mirrors |
| Radiation | Monochromator: graphite / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.05 Å / Relative weight: 1 |
| Reflection | Resolution: 2.7→20 Å / Num. all: 295654 / Num. obs: 178808 / % possible obs: 93.2 % / Observed criterion σ(F): -3 / Observed criterion σ(I): -3 / Redundancy: 1.65 % / Rsym value: 0.066 |
| Reflection shell | Resolution: 2.7→2.75 Å / Rsym value: 0.263 / % possible all: 83.6 |
| Reflection | *PLUS Lowest resolution: 20 Å / Num. measured all: 295654 / Rmerge(I) obs: 0.066 |
| Reflection shell | *PLUS % possible obs: 83.6 % / Rmerge(I) obs: 0.263 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: direct refinement Starting model: PDB entry 1K32 Resolution: 2.7→6 Å / σ(F): 0 / Stereochemistry target values: Engh & Huber
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.7→6 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| Refinement | *PLUS % reflection Rfree: 8.5 % / Rfactor Rfree: 0.28 | |||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: n_bond_d / Dev ideal: 0.01 |
