解像度: 1.4→1.45 Å / 冗長度: 1.1 % / Rmerge(I) obs: 0.229 / Mean I/σ(I) obs: 2.6 / Num. unique all: 771 / % possible all: 4.4
反射
*PLUS
Num. obs: 105327 / Num. measured all: 290225
反射 シェル
*PLUS
最高解像度: 1.4 Å / % possible obs: 0.044 %
-
解析
ソフトウェア
名称
分類
DENZO
データ削減
SCALEPACK
データスケーリング
CNS
精密化
CNS
位相決定
精密化
構造決定の手法: 分子置換 / 解像度: 1.4→29.16 Å / Rfactor Rfree error: 0.002 / Isotropic thermal model: RESTRAINED / 交差検証法: THROUGHOUT / σ(F): 0 / 立体化学のターゲット値: Engh & Huber 詳細: Model refined against first crystal (CHESS) data then later extended to higher resolution with inclusion of second data set (Rfree reflections kept consistent with addition of higher resolution data).
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報
 Methanocaldococcus jannaschii (メタン生成菌)
Methanocaldococcus jannaschii (メタン生成菌) X線回折 /
X線回折 /  データ登録者
データ登録者 引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード









 PDBj
PDBj 集合体
集合体



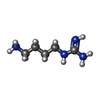
 分子量: 130.191 Da / 分子数: 6 / 由来タイプ: 合成 / 式: C5H14N4
分子量: 130.191 Da / 分子数: 6 / 由来タイプ: 合成 / 式: C5H14N4
 分子量: 118.174 Da / 分子数: 8 / 由来タイプ: 合成 / 式: C6H14O2 / コメント: 沈殿剤*YM
分子量: 118.174 Da / 分子数: 8 / 由来タイプ: 合成 / 式: C6H14O2 / コメント: 沈殿剤*YM 分子量: 18.015 Da / 分子数: 736 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 736 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製
 解析
解析