 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1n02: Solution Structure of a Circular-Permuted Variant of the Potent H... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1n02 | ||||||
|---|---|---|---|---|---|---|---|
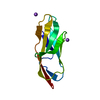
| Title | Solution Structure of a Circular-Permuted Variant of the Potent HIV-inactivating Protein Cyanovirin-N | ||||||
 Components Components | Cyanovirin-N | ||||||
 Keywords Keywords | Viral protein inhibitor / VIRUS/VIRAL PROTEIN INHIBITOR | ||||||
| Function / homology |  Function and homology information Function and homology informationoligosaccharide binding / regulation of defense response to virus / carbohydrate binding Similarity search - Function | ||||||
| Biological species |  Nostoc ellipsosporum (bacteria) Nostoc ellipsosporum (bacteria) | ||||||
| Method | SOLUTION NMR / for dyana, Standard target function-simulated annealing protocol. For CNS, Simulated-annealing in Cartesian space | ||||||
| Model type details | minimized average | ||||||
 Authors Authors | Barrientos, L.G. / Gronenborn, A.M. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2003 Title: Solution Structure of a Circular-Permuted Variant of the Potent HIV-inactivating Protein Cyanovirin-N: Structural Basis for Protein Stability and Oligosaccharide Interaction Authors: Barrientos, L.G. / Louis, J.M. / Ratner, D.M. / Seeberger, P.H. / Gronenborn, A.M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1n02.cif.gz | 770.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1n02.ent.gz | 647.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1n02.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/n0/1n02ftp://data.pdbj.org/pub/pdb/validation_reports/n0/1n02 | HTTPS FTP |
|---|
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data | |
| Other databases |
|










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| NMR ensembles |
|
-Components
| #1: Protein | Mass: 11079.145 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Details: circular-permuted variant / Source: (gene. exp.) Nostoc ellipsosporum (bacteria) / Production host: |
|---|---|
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: SOLUTION NMR | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NMR experiment |
| ||||||||||||||||||||||||||||||||||||||||
| NMR details | Text: The minimized average structure is model 1. Models 2-26 are the the final 25 conformer ensemble. DYANA and CNS were used for refinement. VIRTUALLY COMPLETE RESONANCE ASSIGNMENTS WERE REPORTED I ...Text: The minimized average structure is model 1. Models 2-26 are the the final 25 conformer ensemble. DYANA and CNS were used for refinement. VIRTUALLY COMPLETE RESONANCE ASSIGNMENTS WERE REPORTED I (2001) J.Biom.NMR, 19: 289-90; http://www.bmrb.wisc.edu. Stereo-specific assignments for all the methyl groups of of the eight Leu residues and for the alpha-methylene pr eight Gly, 46 beta-methylene protons and the gamma-methy of the seven Ile were available. In total, 1879 experim were employed, representing ~19 constraints per residue. structures was first calculated with DYANA, based on 114 92 hydrogen bond distances and 193 dihedral angles. Ref values yielded an esemble of 50 structures exhibiting at 0.26+/-0.06 A and 0.64+/-0.08A with respect to the mean backbone (N,CA,C') and all heavy atoms, respectively, an 0.81+/-0.10A^2. This initial ensemble of structures was residual dipolar couplings (80) and chemical shifts (291 programs CNS. Overall, excellent agreement with the exp good covalent geometry was maintained throughout. The f was selected and are presented here as MODELS2-26, and a mean structure of this ensemble is presented here as MOD. |
 HSQC
HSQC- Sample preparation
Sample preparation
| Sample conditions | pH: 6 / Temperature: 300 K |
|---|---|
| Crystal grow | *PLUS Method: other / Details: NMR |
-NMR measurement
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Radiation wavelength | Relative weight: 1 | |||||||||||||||||||||||||
| NMR spectrometer |
|
- Processing
Processing
| NMR software |
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method: for dyana, Standard target function-simulated annealing protocol. For CNS, Simulated-annealing in Cartesian space Software ordinal: 1 | ||||||||||||
| NMR representative | Selection criteria: minimized average structure | ||||||||||||
| NMR ensemble | Conformers submitted total number: 26 |
