 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1myz | ||||||
|---|---|---|---|---|---|---|---|
| Title | CO COMPLEX OF MYOGLOBIN MB-YQR AT RT SOLVED FROM LAUE DATA. | ||||||
 Components Components | Myoglobin | ||||||
 Keywords Keywords | OXYGEN STORAGE/TRANSPORT / OXYGEN STORAGE / CO COMPLEX / RESPIRATORY PROTEIN / HEME / OXYGEN STORAGE-TRANSPORT COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationnitrite reductase activity / Oxidoreductases; Acting on other nitrogenous compounds as donors / sarcoplasm / Oxidoreductases; Acting on a peroxide as acceptor; Peroxidases / removal of superoxide radicals / oxygen carrier activity / peroxidase activity / oxygen binding / heme binding / extracellular exosome / metal ion binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / Rigid Body from starting model / Resolution: 1.6 Å X-RAY DIFFRACTION / SYNCHROTRON / Rigid Body from starting model / Resolution: 1.6 Å | ||||||
 Authors Authors | Bourgeois, D. / Vallone, B. / Schotte, F. / Arcovito, A. / Miele, A.E. / Sciara, G. / Wulff, M. / Anfinrud, P. / Brunori, M. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 2003 Title: Complex landscape of protein structural dynamics unveiled by nanosecond Laue crystallography. Authors: Bourgeois, D. / Vallone, B. / Schotte, F. / Arcovito, A. / Miele, A.E. / Sciara, G. / Wulff, M. / Anfinrud, P. / Brunori, M. #1: Journal: Biophys.J. / Year: 1999Title: Structural dynamics of ligand diffusion in the protein matrix: A study on a new myoglobin mutant Y(B10) Q(E7) R(E10) Authors: Brunori, M. / Cutruzzol, F. / Savino, C. / Travaglini-Allocatelli, C. / Vallone, B. / Gibson, Q.H. #2: Journal: Proc.Natl.Acad.Sci.USA / Year: 2000Title: The role of cavities in protein dynamics: crystal structure of a photolytic intermediate of a mutant myoglobin. Authors: Brunori, M. / Vallone, B. / Cutruzzol, F. / Travaglini-Allocatelli, C. / Berendzen, J. / Chu, K. / Sweet, R.M. / Schlichting, I. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1myz.cif.gz | 52.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1myz.ent.gz | 37.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1myz.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/my/1myzftp://data.pdbj.org/pub/pdb/validation_reports/my/1myz | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1mz0C  100kS  1dxcS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj









- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 17461.250 Da / Num. of mol.: 1 / Mutation: L29Y, H64Q, T67R Source method: isolated from a genetically manipulated source Source: (gene. exp.)  Keywords: L29(B10)Y, H64(E7)Q, T67(E10)R / References: UniProt: P02185 Keywords: L29(B10)Y, H64(E7)Q, T67(E10)R / References: UniProt: P02185 | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical |   Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#3: Chemical | ChemComp-HEM / |   Mass: 616.487 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H32FeN4O4 Mass: 616.487 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H32FeN4O4#4: Chemical | ChemComp-CMO / | 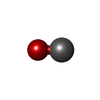  Mass: 28.010 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CO Mass: 28.010 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CO#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 143 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 143 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 10 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.14 Å3/Da / Density % sol: 60.85 % | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 294 K / pH: 8.7 Details: CO-saturated, 2.8M ammonium sulphate, 100mM Tris-Cl, 1 mM dithionite, crystallized in seeded batch, pH 8.7, temperature 294.0K | ||||||||||||||||||||
| Crystal grow | *PLUS pH: 9 / Method: batch methodDetails: Phillips, G.N., (1990) Proteins Struct. Funct. Genet., 7, 358. | ||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 283 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID09 / Beamline: ID09 |
| Detector | Type: MAR CCD 130 mm / Detector: CCD / Date: Dec 18, 2001 |
| Radiation | Protocol: LAUE / Monochromatic (M) / Laue (L): L / Scattering type: neutron |
| Radiation wavelength | Relative weight: 1 |
| Reflection | Resolution: 1.55→29.88 Å / Num. all: 31195 / Num. obs: 31195 / % possible obs: 96 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -3 / Redundancy: 26.7 % / Biso Wilson estimate: 19.7 Å2 / Rsym value: 0.064 / Net I/σ(I): 37.1 |
| Reflection shell | Resolution: 1.55→1.62 Å / Redundancy: 4.1 % / Mean I/σ(I) obs: 4.5 / Num. unique all: 3574 / Rsym value: 0.414 / % possible all: 75.9 |
| Reflection | *PLUS Num. measured all: 832442 / Rmerge(I) obs: 0.064 |
| Reflection shell | *PLUS % possible obs: 75.9 % / Rmerge(I) obs: 0.414 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: Rigid Body from starting model Starting model: 1DXC (MbCO-YQR at 100K) Resolution: 1.6→29.88 Å / Rfactor Rfree error: 0.005 / Data cutoff high absF: 1532735.67 / Data cutoff high rms absF: 1532735.67 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber Details: Sulfate 501 is on the 3 fold axis. Atoms S and O1 seat right on the axis and are given an occupancy of 0.33. Atoms O3 and O4 are generated by symmetry from atom O2. Unobserved (disordered) ...Details: Sulfate 501 is on the 3 fold axis. Atoms S and O1 seat right on the axis and are given an occupancy of 0.33. Atoms O3 and O4 are generated by symmetry from atom O2. Unobserved (disordered) atoms were removed from the ATOM list: 44 ASP OD2 62 LYS CE 62 LYS NZ 96 LYS CE 96 LYS NZ 98 LYS CD 98 LYS CE 98 LYS NZ 102 LYS CD 102 LYS CE 102 LYS NZ 140 LYS CD 140 LYS CE 140 LYS NZ 147 LYS CD 147 LYS CE 147 LYS NZ
| ||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 89.0488 Å2 / ksol: 0.359328 e/Å3 | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 19.4 Å2
| ||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.6→29.88 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.6→1.7 Å / Rfactor Rfree error: 0.014 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS % reflection Rfree: 5 % | ||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
