 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1mxf: Crystal Structure of Inhibitor Complex of Putative Pteridine Redu... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1mxf | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of Inhibitor Complex of Putative Pteridine Reductase 2 (PTR2) from Trypanosoma cruzi | ||||||
 Components Components | PTERIDINE REDUCTASE 2 | ||||||
 Keywords Keywords | OXIDOREDUCTASE / SDR TOPOLOGY / PROTEIN-INHIBITOR COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.3 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.3 Å | ||||||
 Authors Authors | Schormann, N. / Pal, B. / Senkovich, O. / Carson, M. / Howard, A. / Smith, C. / Delucas, L. / Chattopadhyay, D. | ||||||
 Citation Citation | Journal: J.Struct.Biol. / Year: 2005 Title: Crystal structure of Trypanosoma cruzi pteridine reductase 2 in complex with a substrate and an inhibitor. Authors: Schormann, N. / Pal, B. / Senkovich, O. / Carson, M. / Howard, A. / Smith, C. / Delucas, L. / Chattopadhyay, D. #1: Journal: Acta Crystallogr.,Sect.D / Year: 2001Title: Expression, purification, crystallization and preliminary crystallographic analysis of recombinant pteridine reductase of Trypanosoma cruzi Authors: Schormann, N. / Pal, B. / Chattopadhyay, D. | ||||||
| History |
| ||||||
| Remark 999 | sequence the sequence of this molecule is not yet deposited in any sequence database |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1mxf.cif.gz | 204.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1mxf.ent.gz | 166.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1mxf.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/mx/1mxfftp://data.pdbj.org/pub/pdb/validation_reports/mx/1mxf | HTTPS FTP |
|---|
-Related structure data













-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 29321.957 Da / Num. of mol.: 4 / Fragment: pteridine reductase Source method: isolated from a genetically manipulated source Source: (gene. exp.)  #2: Chemical | ChemComp-NDP / 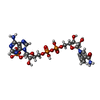  Mass: 745.421 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C21H30N7O17P3 Mass: 745.421 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C21H30N7O17P3#3: Chemical | ChemComp-MTX / 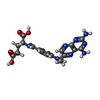  Mass: 454.439 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C20H22N8O5 Mass: 454.439 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C20H22N8O5#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 346 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 346 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.3 Å3/Da / Density % sol: 46.7 % | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / pH: 6.3 Details: Sodium acetate, cacodylate buffer, pH 6.30, temperature 293K | |||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 293 K / pH: 8 / Method: vapor diffusion, hanging dropDetails: Schormann, N., (2001) Acta Crystallogr.,Sect.D, 57, 1671. | |||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 103 K | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 17-ID / Wavelength: 0.9537, 0.9791, 0.9792 / Beamline: 17-ID / Wavelength: 0.9537, 0.9791, 0.9792 | ||||||||||||
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Dec 20, 2000 / Details: MIRRORS | ||||||||||||
| Radiation | Monochromator: GRAPHITE / Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||
| Radiation wavelength |
| ||||||||||||
| Reflection | Resolution: 1.76→36 Å / Num. obs: 89619 / % possible obs: 80 % / Observed criterion σ(I): 0 / Redundancy: 5.1 % / Biso Wilson estimate: 22.6 Å2 / Rmerge(I) obs: 0.037 / Net I/σ(I): 11.8 | ||||||||||||
| Reflection | *PLUS Highest resolution: 2.3 Å / Lowest resolution: 50 Å / Num. obs: 48846 / % possible obs: 97.1 % / Redundancy: 2.9 % / Rmerge(I) obs: 0.084 | ||||||||||||
| Reflection shell | *PLUS Highest resolution: 2.3 Å / Lowest resolution: 2.44 Å / % possible obs: 91.9 % / Redundancy: 1.9 % / Num. unique obs: 7734 / Rmerge(I) obs: 0.299 / Mean I/σ(I) obs: 3.2 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 2.3→19.49 Å / Rfactor Rfree error: 0.005 / Data cutoff high absF: 187450.3 / Data cutoff high rms absF: 187450.3 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 1 / Stereochemistry target values: ENGH & HUBER
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 33.982 Å2 / ksol: 0.335 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 36.1 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.3→19.49 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.3→2.44 Å / Rfactor Rfree error: 0.015 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 50 Å / Rfactor Rfree: 0.247 / Rfactor Rwork: 0.204 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.308 / Rfactor Rwork: 0.274 |
