 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1msa: MANNOSE-SPECIFIC AGGLUTININ (LECTIN) FROM SNOWDROP (GALANTHUS NIV... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1msa | ||||||
|---|---|---|---|---|---|---|---|
| Title | MANNOSE-SPECIFIC AGGLUTININ (LECTIN) FROM SNOWDROP (GALANTHUS NIVALIS) BULBS COMPLEXED WITH METHYL-ALPHA-D-MANNOSIDE | ||||||
 Components Components | AGGLUTININ | ||||||
 Keywords Keywords | LECTIN (AGGLUTININ) / METHYL-ALPHA-D-MANNOSIDE | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of defense response to virus / D-mannose binding / defense response / response to virus / extracellular region Similarity search - Function | ||||||
| Biological species |  Galanthus nivalis (common snowdrop) Galanthus nivalis (common snowdrop) | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.29 Å X-RAY DIFFRACTION / Resolution: 2.29 Å | ||||||
 Authors Authors | Wright, C.S. / Hester, G. | ||||||
 Citation Citation | Journal: Nat.Struct.Biol. / Year: 1995 Title: Structure of mannose-specific snowdrop (Galanthus nivalis) lectin is representative of a new plant lectin family. Authors: Hester, G. / Kaku, H. / Goldstein, I.J. / Wright, C.S. #1: Journal: Eur.J.Biochem. / Year: 1991Title: Biosynthesis, Primary Structure and Molecular Cloning of Snowdrop (Galanthus Nivalis) Lectin Authors: Van Damme, E.J.M. / Kaku, H. / Perini, F. / Goldstein, I.J. / Peeters, B. / Yagi, F. / Decock, B. / Peumans, W.J. #2: Journal: J.Biol.Chem. / Year: 1990Title: Crystallization and Preliminary X-Ray Diffraction Results of Snowdrop (Galanthus Nivalis) Lectin Authors: Wright, C.S. / Kaku, H. / Goldstein, I.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1msa.cif.gz | 104.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1msa.ent.gz | 82.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1msa.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ms/1msaftp://data.pdbj.org/pub/pdb/validation_reports/ms/1msa | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|








-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||||||
| Unit cell |
| ||||||||||||||||
| Atom site foot note | 1: GLY A 98 - THR A 99 OMEGA = 359.12 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 2: GLY B 98 - THR B 99 OMEGA = 0.90 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 3: GLY C 98 - THR C 99 OMEGA = 357.74 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 4: GLY D 98 - THR D 99 OMEGA = 0.32 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION | ||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
| ||||||||||||||||
| Details | MTRIX THE TRANSFORMATIONS PRESENTED ON MTRIX RECORDS BELOW DESCRIBE NON-CRYSTALLOGRAPHIC RELATIONSHIPS AMONG THE VARIOUS DOMAINS IN THIS ENTRY. APPLYING THE APPROPRIATE MTRIX TRANSFORMATION TO THE RESIDUES LISTED FIRST WILL YIELD APPROXIMATE COORDINATES FOR THE RESIDUES LISTED SECOND. APPLIED TO TRANSFORMED TO MTRIX RESIDUES RESIDUES RMSD M1 A 1 .. A 109 B 1 .. B 109 0.360 M2 A 1 .. A 109 C 1 .. C 109 0.507 M3 A 1 .. A 109 D 1 .. D 109 0.568 |
-Components
| #1: Protein | Mass: 12061.348 Da / Num. of mol.: 4 / Source method: isolated from a natural source / Source: (natural) Galanthus nivalis (common snowdrop) / References: UniProt: P30617#2: Sugar | ChemComp-MMA / 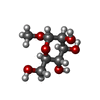  Type: D-saccharide / Mass: 194.182 Da / Num. of mol.: 12 Type: D-saccharide / Mass: 194.182 Da / Num. of mol.: 12Source method: isolated from a genetically manipulated source Formula: C7H14O6 #3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 327 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 327 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | Source details | MOLECULE_NAME: GALANTHUS NIVALIS AGGLUTININ. THE SNOWDROP IS A REPRESENTATIVE OF THE PLANT FAMILY ...MOLECULE_NAME: GALANTHUS NIVALIS AGGLUTININ | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.86 Å3/Da / Density % sol: 57.03 % | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS pH: 8 / Method: unknown | |||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction source | Wavelength: 1.5418 Å |
|---|---|
| Detector | Detector: AREA DETECTOR / Date: Feb 20, 1993 |
| Radiation | Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Num. obs: 23359 / % possible obs: 91.7 % / Observed criterion σ(I): 0 / Redundancy: 3.49 % / Rmerge(I) obs: 0.044 |
| Reflection | *PLUS Highest resolution: 2.295 Å / Rmerge(I) obs: 0.044 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.29→8 Å / σ(F): 0 Details: THREONINE 99 HAS A PEPTIDE TORSION ANGLE WITH A CIS CONFORMATION. IN THE TOPOLOGY AND PARAMETER FILES USED FOR REFINEMENT A NEW RESIDUE CALLED CTH WAS INTRODUCED. THIS IS A THR RESIDUE WITH ...Details: THREONINE 99 HAS A PEPTIDE TORSION ANGLE WITH A CIS CONFORMATION. IN THE TOPOLOGY AND PARAMETER FILES USED FOR REFINEMENT A NEW RESIDUE CALLED CTH WAS INTRODUCED. THIS IS A THR RESIDUE WITH A NEW ATOM TYPE BEING A MAIN CHAIN NITROGEN ATOM WHICH IS INVOLVED IN A CIS PEPTIDE BOND.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 18.15 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.22 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.29→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.295 Å / Rfactor Rfree: 0.24 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: x_dihedral_angle_d / Dev ideal: 27.24 |
