 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
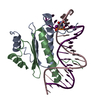
Yorodumi- PDB-1mnm: YEAST MATALPHA2/MCM1/DNA TERNARY TRANSCRIPTION COMPLEX CRYSTAL ST... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1mnm | ||||||
|---|---|---|---|---|---|---|---|
| Title | YEAST MATALPHA2/MCM1/DNA TERNARY TRANSCRIPTION COMPLEX CRYSTAL STRUCTURE | ||||||
 Components Components |
| ||||||
 Keywords Keywords | TRANSCRIPTION/DNA / TRANSCRIPTION REGULATION / TRANSCRIPTIONAL REPRESSION / DNA-BINDING PROTEIN / COMPLEX (TRANSCRIPTION-HOMEOBOX-DNA) / TRANSCRIPTION-DNA COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationRNA polymerase II cis-regulatory region sequence-specific DNA binding, bending / positive regulation of arginine biosynthetic process / NGF-stimulated transcription / regulation of mating type switching / negative regulation of arginine catabolic process / : / regulation of mating-type specific transcription, DNA-templated / : / RNA polymerase II transcription repressor complex / arginine metabolic process ...RNA polymerase II cis-regulatory region sequence-specific DNA binding, bending / positive regulation of arginine biosynthetic process / NGF-stimulated transcription / regulation of mating type switching / negative regulation of arginine catabolic process / : / regulation of mating-type specific transcription, DNA-templated / : / RNA polymerase II transcription repressor complex / arginine metabolic process / DNA binding, bending / DNA replication origin binding / molecular sequestering activity / DNA-binding transcription repressor activity, RNA polymerase II-specific / G2/M transition of mitotic cell cycle / RNA polymerase II transcription regulator complex / DNA-binding transcription activator activity, RNA polymerase II-specific / sequence-specific DNA binding / RNA polymerase II-specific DNA-binding transcription factor binding / DNA-binding transcription factor activity, RNA polymerase II-specific / protein dimerization activity / RNA polymerase II cis-regulatory region sequence-specific DNA binding / regulation of transcription by RNA polymerase II / chromatin / negative regulation of transcription by RNA polymerase II / positive regulation of transcription by RNA polymerase II / nucleus / cytosol Similarity search - Function | ||||||
| Biological species |  synthetic construct (others) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.25 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.25 Å | ||||||
 Authors Authors | Tan, S. / Richmond, T.J. | ||||||
 Citation Citation | Journal: Nature / Year: 1998 Title: Crystal structure of the yeast MATalpha2/MCM1/DNA ternary complex. Authors: Tan, S. / Richmond, T.J. #1: Journal: Nature / Year: 1995Title: Structure of Serum Response Factor Core Bound to DNA Authors: Pellegrini, L. / Tan, S. / Richmond, T.J. #2: Journal: Cell(Cambridge,Mass.) / Year: 1991Title: Crystal Structure of a MAT Alpha 2 Homeodomain-Operator Complex Suggests a General Model for Homeodomain-DNA Interactions Authors: Wolberger, C. / Vershon, A.K. / Liu, B. / Johnson, A.D. / Pabo, C.O. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1mnm.cif.gz | 101.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1mnm.ent.gz | 77.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1mnm.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/mn/1mnmftp://data.pdbj.org/pub/pdb/validation_reports/mn/1mnm | HTTPS FTP |
|---|
-Related structure data










-Links
 PDBj
PDBj






































- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: DNA chain | Mass: 8019.217 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) synthetic construct (others) | ||||
|---|---|---|---|---|---|
| #2: DNA chain | Mass: 7952.150 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) synthetic construct (others) | ||||
| #3: Protein | Mass: 11422.067 Da / Num. of mol.: 2 / Fragment: RESIDUES 1 - 100 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Production host:  Keywords: FRAGMENT: RESIDUES 1 - 100 / References: UniProt: P11746 Keywords: FRAGMENT: RESIDUES 1 - 100 / References: UniProt: P11746#4: Protein | Mass: 10315.832 Da / Num. of mol.: 2 / Fragment: RESIDUES 113 - 189 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Production host: Keywords: FRAGMENT: RESIDUES 113 - 189 / References: UniProt: Q6B2C0, UniProt: P0CY08*PLUS#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 53 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 53 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.22 Å3/Da / Density % sol: 61.75 % Description: ADDITIONAL LOW RESOLUTION DATA SET COLLECTED USING ROTATING ANODE GENERATOR. |
|---|---|
| Crystal grow | pH: 5.5 / Details: pH 5.50 |
| Crystal grow | *PLUS Method: unknown |
-Data collection
| Diffraction | Mean temperature: 110 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: BM14 / Beamline: BM14 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Sep 1, 1996 / Details: MIRRORS |
| Radiation | Monochromator: SI(111) / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Relative weight: 1 |
| Reflection | Resolution: 2.25→20 Å / Num. obs: 35613 / % possible obs: 94.6 % / Biso Wilson estimate: 38.2 Å2 / Rmerge(I) obs: 0.065 / Net I/σ(I): 9.8 |
| Reflection shell | Resolution: 2.25→2.29 Å / Rmerge(I) obs: 0.364 / Mean I/σ(I) obs: 1.7 / % possible all: 0.9 |
| Reflection | *PLUS Highest resolution: 2.25 Å / Lowest resolution: 20 Å / % possible obs: 94.6 % / Num. measured all: 327727 |
| Reflection shell | *PLUS Highest resolution: 2.25 Å / Lowest resolution: 2.29 Å / Mean I/σ(I) obs: 1.7 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1SRS AND PDB ENTRY 1APL Resolution: 2.25→25 Å / Rfactor Rfree error: 0.7 / Data cutoff high absF: 10000000 / Data cutoff low absF: 0.001 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 2 Details: BULK SOLVENT MODEL USED IN X-PLOR. TNT REFINEMENT PACKAGE ALSO USED.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 52.5 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.25→25 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.25→2.39 Å / Rfactor Rfree error: 0.02 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.843 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.25 Å / Lowest resolution: 25 Å / Num. reflection obs: 34859 / σ(F): 2 / % reflection Rfree: 5 % / Rfactor obs: 0.24 / Rfactor Rwork: 0.24 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 52.5 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.36 / % reflection Rfree: 5.2 % |
