 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 1lwd | ||||||
|---|---|---|---|---|---|---|---|
| タイトル | CRYSTAL STRUCTURE OF NADP-DEPENDENT ISOCITRATE DEHYDROGENASE FROM PORCINE HEART MITOCHONDRIA | ||||||
 要素 要素 | Isocitrate Dehydrogenase | ||||||
 キーワード キーワード | OXIDOREDUCTASE / TRICARBOXYLIC ACID CYCLE / NADP | ||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報isocitrate metabolic process / isocitrate dehydrogenase (NADP+) / isocitrate dehydrogenase (NADP+) activity / 2-oxoglutarate metabolic process / NADP metabolic process / glyoxylate cycle / tricarboxylic acid cycle / NAD binding / magnesium ion binding / mitochondrion 類似検索 - 分子機能 | ||||||
| 生物種 |  | ||||||
| 手法 |  X線回折 / 多波長異常分散 / 解像度: 1.85 Å X線回折 / 多波長異常分散 / 解像度: 1.85 Å | ||||||
 データ登録者 データ登録者 | Ceccarelli, C. / Bahnson, B.J. | ||||||
 引用 引用 | ジャーナル: J.Biol.Chem. / 年: 2002 タイトル: Crystal Structure of Porcine Mitochondrial NADP+-Dependent Isocitrate Dehydrogenase Complexed with Mn2+ and Isocitrate 著者: Ceccarelli, C. / Grodsky, N.B. / Ariyaratne, N. / Colman, R.F. / Bahnson, B.J. | ||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 1lwd.cif.gz | 191.1 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb1lwd.ent.gz | 150.8 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 1lwd.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| 文書・要旨 | 1lwd_validation.pdf.gz | 463.4 KB | 表示 | wwPDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | 1lwd_full_validation.pdf.gz | 473.1 KB | 表示 | |
| XML形式データ | 1lwd_validation.xml.gz | 38.1 KB | 表示 | |
| CIF形式データ | 1lwd_validation.cif.gz | 56.9 KB | 表示 | |
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/lw/1lwdftp://data.pdbj.org/pub/pdb/validation_reports/lw/1lwd | HTTPS FTP |
-関連構造データ










-リンク
 PDBj
PDBj


- 集合体
集合体
| 登録構造単位 | 
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||||||||
| 単位格子 |
| |||||||||||||||
| Components on special symmetry positions |
| |||||||||||||||
| 非結晶学的対称性 (NCS) | NCS oper: (Code: given Matrix: (-0.53688, 0.63102, 0.55998), ベクター: 詳細 | THE BIOLOGICALLY FUNCTIONAL MOLECULE IS A DIMER FORMED BY SUBUNITS A AND B, EACH OF WHICH IS PRESENT IN THE CRYSTALLOGRAPHIC ASYMMETRIC UNIT | |
-要素
| #1: タンパク質 | 分子量: 46695.219 Da / 分子数: 2 / 由来タイプ: 組換発現 / 由来: (組換発現)  #2: 化合物 |   分子量: 54.938 Da / 分子数: 2 / 由来タイプ: 合成 / 式: Mn 分子量: 54.938 Da / 分子数: 2 / 由来タイプ: 合成 / 式: Mn#3: 化合物 | 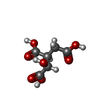  分子量: 192.124 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C6H8O7 分子量: 192.124 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C6H8O7#4: 化合物 |   分子量: 96.063 Da / 分子数: 2 / 由来タイプ: 合成 / 式: SO4 分子量: 96.063 Da / 分子数: 2 / 由来タイプ: 合成 / 式: SO4#5: 水 | ChemComp-HOH / |  分子量: 18.015 Da / 分子数: 715 / 由来タイプ: 天然 / 式: H2O 分子量: 18.015 Da / 分子数: 715 / 由来タイプ: 天然 / 式: H2O |
|---|
-実験情報
-実験
| 実験 | 手法: X線回折 / 使用した結晶の数: 1 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 2.73 Å3/Da / 溶媒含有率: 55 % 解説: THE STRUCTURE OF THE SE-MET CRYSTAL WAS SOLVED BY MULTIWAVELENGTH ANOMALOUS DIFFRACTION (MAD). THE THREE-WAVELENGTH MAD EXPERIMENT WAS PERFORMED ON BEAMLINE X12-C AT THE NSLS, BROOKHAVEN NATIONAL LABORATORY. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 結晶化 | 温度: 277 K / 手法: 蒸気拡散法, ハンギングドロップ法 / pH: 7.7 詳細: 20 MG/ML PROTEIN, 100 MM TRIETHANOLAMINE CHLORIDE PH 7.7, 150 MM SODIUM SULFATE, 8 MM ISOCITRATE, 4 MM MANGANESE SULFATE, 20% PEG 6000, 3% GLYCEROL, pH 7.70, VAPOR DIFFUSION, HANGING DROP, temperature 277K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 結晶化 | *PLUS 温度: 4 ℃ / pH: 7.7 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶液の組成 | *PLUS
|
-データ収集
| 回折 | 平均測定温度: 110 K |
|---|---|
| 放射光源 | 由来: 回転陽極 / タイプ: RIGAKU RUH3R / 波長: 1.5418 / 波長: 1.5418 Å |
| 検出器 | タイプ: RIGAKU RAXIS IV / 検出器: IMAGE PLATE / 日付: 1999年12月1日 / 詳細: OSMIC MIRRORS |
| 放射 | プロトコル: SINGLE WAVELENGTH / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray |
| 放射波長 | 波長: 1.5418 Å / 相対比: 1 |
| 反射 | 解像度: 1.8→30 Å / Num. all: 88689 / Num. obs: 88689 / % possible obs: 95.9 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / 冗長度: 3.767 % / Biso Wilson estimate: 22.9 Å2 / Rmerge(I) obs: 0.073 / Net I/σ(I): 11.6 |
| 反射 シェル | 解像度: 1.8→1.86 Å / 冗長度: 3.643 % / Rmerge(I) obs: 0.963 / Mean I/σ(I) obs: 1.4 / Num. unique all: 8584 / % possible all: 92.7 |
| 反射 | *PLUS 最高解像度: 1.8 Å / 最低解像度: 30 Å / Num. measured all: 334110 |
- 解析
解析
| ソフトウェア |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 構造決定の手法: 多波長異常分散 開始モデル: ISOMORPHOUS SE-MET CRYSTAL STRUCTURE 解像度: 1.85→29.23 Å / Rfactor Rfree error: 0.002 / Isotropic thermal model: RESTRAINED / 交差検証法: THROUGHOUT / σ(F): 0 / 立体化学のターゲット値: Engh & Huber 詳細: NCS RESTRAINTS WERE APPLIED TO 3074 OF 3285 PAIRS OF ATOMS IN THE A AND B SUBUNITS OF THE PROTEIN. SEE REMARK 295 FOR RESIDUES AND SIDE CHAIN ATOMS OMITTED FROM NCS RESTRAINTS.
| ||||||||||||||||||||||||||||||||||||
| 溶媒の処理 | 溶媒モデル: FLAT MODEL / Bsol: 46.6925 Å2 / ksol: 0.352708 e/Å3 | ||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | Biso mean: 27.4 Å2
| ||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||
| 精密化ステップ | サイクル: LAST / 解像度: 1.85→29.23 Å
| ||||||||||||||||||||||||||||||||||||
| 拘束条件 |
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | NCS model details: RESTRAINED / Rms dev Biso : 1.936 Å2 / Rms dev position: 0.066 Å / Weight Biso : 2 / Weight position: 50 | ||||||||||||||||||||||||||||||||||||
| LS精密化 シェル | 解像度: 1.85→1.97 Å / Rfactor Rfree error: 0.009 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||
| 精密化 | *PLUS % reflection Rfree: 10 % / Rfactor Rfree: 0.21 | ||||||||||||||||||||||||||||||||||||
| 溶媒の処理 | *PLUS | ||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | *PLUS | ||||||||||||||||||||||||||||||||||||
| 拘束条件 | *PLUS
|
