 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1lth: T AND R STATES IN THE CRYSTALS OF BACTERIAL L-LACTATE DEHYDROGENA... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1lth | ||||||
|---|---|---|---|---|---|---|---|
| Title | T AND R STATES IN THE CRYSTALS OF BACTERIAL L-LACTATE DEHYDROGENASE REVEAL THE MECHANISM FOR ALLOSTERIC CONTROL | ||||||
 Components Components | L-LACTATE DEHYDROGENASE (T- AND R- STATE TETRAMER COMPLEX) | ||||||
 Keywords Keywords | OXIDOREDUCTASE / CHOH(D)-NAD(A) / ALLOSTERIC ENZYME | ||||||
| Function / homology |  Function and homology information Function and homology informationL-lactate dehydrogenase / L-lactate dehydrogenase (NAD+) activity / lactate metabolic process / glycolytic process / cytoplasm Similarity search - Function | ||||||
| Biological species |  Bifidobacterium longum subsp. longum (bacteria) Bifidobacterium longum subsp. longum (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.5 Å X-RAY DIFFRACTION / Resolution: 2.5 Å | ||||||
 Authors Authors | Iwata, S. / Ohta, T. | ||||||
 Citation Citation | Journal: Nat.Struct.Biol. / Year: 1994 Title: T and R states in the crystals of bacterial L-lactate dehydrogenase reveal the mechanism for allosteric control. Authors: Iwata, S. / Kamata, K. / Yoshida, S. / Minowa, T. / Ohta, T. #1: Journal: J.Mol.Biol. / Year: 1994Title: A Regular 1:1 Complex of Two Allosteric States in the Single Crystal of L-Lactate Dehydrogenase from Bifidobacterium Longum Authors: Iwata, S. / Yoshida, S. / Ohta, T. #2: Journal: J.Mol.Biol. / Year: 1993Title: Molecular Basis of Allosteric Activation of Bacterial L-Lactate Dehydrogenase Authors: Iwata, S. / Ohta, T. #3: Journal: FARADAY DISC.CHEM.SOC / Year: 1992Title: Mechanism of Allosteric Transition of Bacterial L-Lactate Dehydrogenase Authors: Ohta, T. / Minowa, T. / Yokota, K. / Iwata, S. #4: Journal: J.Biochem.(Tokyo) / Year: 1989Title: Crystallization of and Preliminary Crystallographic Data for Allosteric L-Lactate Dehydrogenase from Bifidobacterium Longum Authors: Iwata, S. / Minowa, T. / Mikani, B. / Morita, Y. / Ohta, T. #5: Journal: Gene / Year: 1989Title: Sequence and Characteristics of the Bifidobacterium Longum Gene Encoding L-Lactate Dehydrogenase and the Primary Structure of the Enzyme: A New Feature of the Allosteric Site Authors: Minowa, T. / Iwata, S. / Sakai, H. / Masaki, H. / Ohta, T. #6: Journal: Agric.Biol.Chem. / Year: 1989Title: Amino Acid Residues in the Allosteric Site of L-Lactate Dehydrogenase from Bifidobacterium Longum Authors: Iwata, S. / Minowa, T. / Sakai, H. / Ohta, T. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1lth.cif.gz | 138.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1lth.ent.gz | 109.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1lth.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/lt/1lthftp://data.pdbj.org/pub/pdb/validation_reports/lt/1lth | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|









-Links
 PDBj
PDBj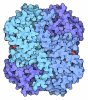



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Atom site foot note | 1: CIS PROLINE - PRO T 126 / 2: CIS PROLINE - PRO R 126 | ||||||||
| Details | THIS CRYSTAL STRUCTURE REFERENCES THE REGULAR 1:1 COMPLEX OF T-STATE (LOW AFFINITY TO SUBSTRATE) AND R-STATE (HIGH AFFINITY TO SUBSTRATE) TETRAMERS OF L-LACTATE DEHYDROGENASE FROM BIFIDOBACTERIUM LONGUM. THE CRYSTALLOGRAPHIC ASYMMETRIC UNIT CONTAINS ONE T-STATE SUBUNIT AND ONE R-STATE SUBUNIT ASSIGNED CHAIN IDENTIFIERS "T" AND "R", RESPECTIVELY. SYMMETRY THE CRYSTALLOGRAPHIC SYMMETRY TRANSFORMATIONS PRESENTED BELOW GENERATE THE SUBUNITS OF THE POLYMERIC MOLECULE. APPLIED TO RESIDUES: T 6 .. T 321 FORMATION OF THE P-AXIS RELATED SUBUNIT OF THE T-STATE TETRAMER SYMMETRY1 1 1.000000 0.000000 0.000000 0.00000 SYMMETRY2 1 0.000000 -1.000000 0.000000 0.00000 SYMMETRY3 1 0.000000 0.000000 -1.000000 0.00000 APPLIED TO RESIDUES: T 6 .. T 321 FORMATION OF THE Q-AXIS RELATED SUBUNIT OF THE T-STATE TETRAMER SYMMETRY1 2 -1.000000 0.000000 0.000000 0.00000 SYMMETRY2 2 0.000000 1.000000 0.000000 0.00000 SYMMETRY3 2 0.000000 0.000000 -1.000000 0.00000 APPLIED TO RESIDUES: T 6 .. T 321 FORMATION OF THE R-AXIS RELATED SUBUNIT OF THE T-STATE TETRAMER SYMMETRY1 3 -1.000000 0.000000 0.000000 0.00000 SYMMETRY2 3 0.000000 -1.000000 0.000000 0.00000 SYMMETRY3 3 0.000000 0.000000 1.000000 0.00000 APPLIED TO RESIDUES: R 6 .. R 322 FORMATION OF THE P-AXIS RELATED SUBUNIT OF THE R-STATE TETRAMER SYMMETRY1 4 1.000000 0.000000 0.000000 0.00000 SYMMETRY2 4 0.000000 -1.000000 0.000000 147.92900 SYMMETRY3 4 0.000000 0.000000 -1.000000 35.46099 APPLIED TO RESIDUES: R 6 .. R 322 FORMATION OF THE Q-AXIS RELATED SUBUNIT OF THE R-STATE TETRAMER SYMMETRY1 5 -1.000000 0.000000 0.000000 74.18398 SYMMETRY2 5 0.000000 1.000000 0.000000 0.00000 SYMMETRY3 5 0.000000 0.000000 -1.000000 35.46099 APPLIED TO RESIDUES: R 6 .. R 322 FORMATION OF THE R-AXIS RELATED SUBUNIT OF THE R-STATE TETRAMER SYMMETRY1 6 -1.000000 0.000000 0.000000 74.18398 SYMMETRY2 6 0.000000 -1.000000 0.000000 147.92900 SYMMETRY3 6 0.000000 0.000000 1.000000 0.00000 |
-Components
| #1: Protein | Mass: 34128.777 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Bifidobacterium longum subsp. longum (bacteria)Species: Bifidobacterium longum / Strain: subsp. longum / Gene: BIFIDOBACTERIUM LONGUM LDH GENE / Plasmid: PUBM9 (A DERIVATIVE OF PUC119) / Production host: References: UniProt: P19869, UniProt: E8ME30*PLUS, L-lactate dehydrogenase #2: Sugar | 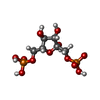  Type: D-saccharide, beta linking / Mass: 340.116 Da / Num. of mol.: 2 Type: D-saccharide, beta linking / Mass: 340.116 Da / Num. of mol.: 2Source method: isolated from a genetically manipulated source Formula: C6H14O12P2 #3: Chemical |   Mass: 663.425 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H27N7O14P2 / Comment: NAD*YM Mass: 663.425 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H27N7O14P2 / Comment: NAD*YM#4: Chemical | ChemComp-OXM / |   Mass: 89.050 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H3NO3 Mass: 89.050 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H3NO3#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 301 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 301 / Source method: isolated from a natural source / Formula: H2OCompound details | THE T-STATE TETRAMERS, THAT BIND NADH AND FBP, ARE CENTERED AT THE ORIGIN AND ITS EQUIVALENTS. THE ...THE T-STATE TETRAMERS, THAT BIND NADH AND FBP, ARE CENTERED AT THE ORIGIN AND ITS EQUIVALENT | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.85 Å3/Da / Density % sol: 56.89 % | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS Temperature: 10 ℃ / Method: vapor diffusion, hanging drop | |||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Radiation | Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| Reflection | Resolution: 2.5→100 Å / Num. obs: 24341 / % possible obs: 89.1 % / Observed criterion σ(I): 1 |
| Reflection | *PLUS Highest resolution: 2.5 Å / Lowest resolution: 100 Å / % possible obs: 97.6 % / Num. measured all: 115542 / Rmerge(I) obs: 0.055 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.5→10 Å / σ(F): 2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 23.79 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.25 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.5→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|
