 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1lrw: Crystal structure of methanol dehydrogenase from P. denitrificans -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1lrw | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of methanol dehydrogenase from P. denitrificans | ||||||
 Components Components |
| ||||||
 Keywords Keywords | OXIDOREDUCTASE / Heavy subunits: 8-fold beta-propeller superbarrel | ||||||
| Function / homology |  Function and homology information Function and homology informationmethanol dehydrogenase (cytochrome c) / methanol oxidation / alcohol dehydrogenase (cytochrome c(L)) activity / pyrroloquinoline quinone binding / methanol metabolic process / alcohol dehydrogenase (NAD+) activity / outer membrane-bounded periplasmic space / periplasmic space / calcium ion binding / membrane Similarity search - Function | ||||||
| Biological species |  Paracoccus denitrificans (bacteria) Paracoccus denitrificans (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.5 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.5 Å | ||||||
 Authors Authors | Xia, Z.-X. / Dai, W.-W. / He, Y.-N. / White, S.A. / Mathews, F.S. / Davidson, V.L. | ||||||
 Citation Citation | Journal: J.Biol.Inorg.Chem. / Year: 2003 Title: X-ray structure of methanol dehydrogenase from Paracoccus denitrificans and molecular modeling of its interactions with cytochrome c-551i Authors: Xia, Z.-X. / Dai, W.-W. / He, Y.-N. / White, S.A. / Mathews, F.S. / Davidson, V.L. | ||||||
| History |
| ||||||
| Remark 999 | sequence authors state that the published sequence of ALA 444- ALA 449, ALA 451 and VAL 490 does ... sequence authors state that the published sequence of ALA 444- ALA 449, ALA 451 and VAL 490 does not match the electron density. Based on the electron density, they established the x-ray sequence Gly 444- Ser 450, Leu 452 and ALA 490 in which Gly 447 is an insertion. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1lrw.cif.gz | 281 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1lrw.ent.gz | 226.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1lrw.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/lr/1lrwftp://data.pdbj.org/pub/pdb/validation_reports/lr/1lrw | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4aahS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 66811.844 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Source: (natural) Paracoccus denitrificans (bacteria) / References: UniProt: P12293, EC: 1.1.99.8#2: Protein | Mass: 9465.473 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Source: (natural) Paracoccus denitrificans (bacteria) / References: UniProt: P29898, EC: 1.1.99.8#3: Chemical |   Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca#4: Chemical | 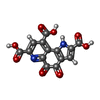  Mass: 330.206 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H6N2O8 Mass: 330.206 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H6N2O8#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 403 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 403 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.44 Å3/Da / Density % sol: 49.53 % | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: seeding / pH: 8.3 Details: PEG 3350, Tris-HCl, Li2SO4, pH 8.3, seeding, temperature 293K | |||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 8.5 / Method: vapor diffusion, hanging drop | |||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 277 K |
|---|---|
| Diffraction source | Wavelength: 1.5418 Å |
| Detector | Detector: AREA DETECTOR |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Highest resolution: 2.49 Å / Num. all: 52799 / Num. obs: 49524 / % possible obs: 93.8 % / Redundancy: 6.4 % / Biso Wilson estimate: 10.8 Å2 / Rsym value: 0.093 / Net I/σ(I): 13.9 |
| Reflection shell | Resolution: 2.49→2.68 Å / Redundancy: 2 % / Mean I/σ(I) obs: 1.9 / Num. unique all: 7330 / Rsym value: 0.262 / % possible all: 70.7 |
| Reflection | *PLUS Lowest resolution: 40 Å / Num. measured all: 317794 / Rmerge(I) obs: 0.093 |
| Reflection shell | *PLUS % possible obs: 70.1 % / Rmerge(I) obs: 0.262 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ID 4AAH Resolution: 2.5→19.76 Å / Rfactor Rfree error: 0.003 / Data cutoff high absF: 53755.76 / Data cutoff high rms absF: 53755.76 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 Details: non-crystallographic symmetry restraint was not applied
| ||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 27.9669 Å2 / ksol: 0.311306 e/Å3 | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 23 Å2
| ||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.5→19.76 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.5→2.59 Å / Rfactor Rfree error: 0.024 / Total num. of bins used: 10
| ||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.49 Å / Lowest resolution: 40 Å / % reflection Rfree: 10 % | ||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Highest resolution: 2.49 Å / Lowest resolution: 2.68 Å |
