 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1lii: STRUCTURE OF T. GONDII ADENOSINE KINASE BOUND TO ADENOSINE 2 AND ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1lii | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | STRUCTURE OF T. GONDII ADENOSINE KINASE BOUND TO ADENOSINE 2 AND AMP-PCP | |||||||||
 Components Components | adenosine kinase | |||||||||
 Keywords Keywords | TRANSFERASE / ALPHA-BETA STRUCTURE | |||||||||
| Function / homology |  Function and homology information Function and homology informationadenosine kinase / adenosine kinase activity / AMP salvage / purine ribonucleoside salvage / purine nucleobase metabolic process / ATP binding / metal ion binding / nucleus / cytosol Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 1.73 Å X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 1.73 Å | |||||||||
 Authors Authors | Schumacher, M.A. / Scott, D.M. / Mathews, I.I. / Ealick, S.E. / Roos, D.S. / Ullman, B. / Brennan, R.G. | |||||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2000 Title: Crystal structures of Toxoplasma gondii adenosine kinase reveal a novel catalytic mechanism and prodrug binding. Authors: Schumacher, M.A. / Scott, D.M. / Mathews, I.I. / Ealick, S.E. / Roos, D.S. / Ullman, B. / Brennan, R.G. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1lii.cif.gz | 82.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1lii.ent.gz | 59.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1lii.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/li/1liiftp://data.pdbj.org/pub/pdb/validation_reports/li/1lii | HTTPS FTP |
|---|
-Related structure data













-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 38373.824 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  |
|---|
-Non-polymers , 5 types, 174 molecules 


| #2: Chemical | ChemComp-MG /  Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg |
|---|---|
| #3: Chemical | ChemComp-CL /  Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl |
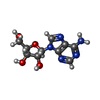
| #4: Chemical | ChemComp-ADN /  Mass: 267.241 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H13N5O4 Mass: 267.241 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H13N5O4 |
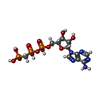
| #5: Chemical | ChemComp-ACP /  Mass: 505.208 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H18N5O12P3 / Comment: AMP-PCP, energy-carrying molecule analogue*YM Mass: 505.208 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H18N5O12P3 / Comment: AMP-PCP, energy-carrying molecule analogue*YM |
| #6: Water | ChemComp-HOH / Mass: 18.015 Da / Num. of mol.: 170 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.29 Å3/Da / Density % sol: 46.23 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: ammonium sulphate, pH 6.50, VAPOR DIFFUSION, HANGING DROP, temperature 298K | ||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 6.5 | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SRS  / Beamline: PX7.2 / Wavelength: 1.08 / Beamline: PX7.2 / Wavelength: 1.08 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Oct 1, 1998 |
| Radiation | Monochromator: graphite / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.08 Å / Relative weight: 1 |
| Reflection | Resolution: 1.72→10 Å / Num. obs: 38408 / % possible obs: 98 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 1 / Redundancy: 4 % / Biso Wilson estimate: 15 Å2 / Rmerge(I) obs: 0.059 / Net I/σ(I): 8.3 |
| Reflection shell | Resolution: 1.72→1.76 Å / Redundancy: 3 % / Rmerge(I) obs: 0.242 / % possible all: 98 |
| Reflection | *PLUS Highest resolution: 1.71 Å / Lowest resolution: 10 Å / % possible obs: 99 % / Num. measured all: 132352 / Rmerge(I) obs: 0.059 |
| Reflection shell | *PLUS % possible obs: 99 % / Rmerge(I) obs: 0.285 / Mean I/σ(I) obs: 2.3 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.73→10 Å / σ(F): 1 / Stereochemistry target values: ENGH & HUBER
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.73→10 Å
| |||||||||||||||||||||||||
| Refinement | *PLUS % reflection Rfree: 10 % / Rfactor obs: 0.178 / Rfactor Rfree: 0.229 / Rfactor Rwork: 0.178 / Highest resolution: 1.71 Å | |||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
