 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1lie: X-RAY CRYSTALLOGRAPHIC STRUCTURES OF ADIPOCYTE LIPID BINDING PROT... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1lie | ||||||
|---|---|---|---|---|---|---|---|
| Title | X-RAY CRYSTALLOGRAPHIC STRUCTURES OF ADIPOCYTE LIPID BINDING PROTEIN COMPLEXED WITH PALMITATE AND HEXADECANESULFONIC ACID. PROPERTIES OF CAVITY BINDING SITES | ||||||
 Components Components | ADIPOCYTE LIPID-BINDING PROTEIN | ||||||
 Keywords Keywords | LIPID BINDING PROTEIN / LIPID-BINDING PROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology informationlipase activator activity / Triglyceride catabolism / lipid droplet formation / triglyceride catabolic process / hormone receptor binding / long-chain fatty acid transmembrane transporter activity / cellular response to lithium ion / long-chain fatty acid binding / white fat cell differentiation / long-chain fatty acid transport ...lipase activator activity / Triglyceride catabolism / lipid droplet formation / triglyceride catabolic process / hormone receptor binding / long-chain fatty acid transmembrane transporter activity / cellular response to lithium ion / long-chain fatty acid binding / white fat cell differentiation / long-chain fatty acid transport / fatty acid transport / negative regulation of protein kinase activity / lipid droplet / response to bacterium / brown fat cell differentiation / cholesterol homeostasis / fatty acid metabolic process / fatty acid binding / cellular response to tumor necrosis factor / positive regulation of inflammatory response / positive regulation of cold-induced thermogenesis / negative regulation of DNA-templated transcription / positive regulation of cell population proliferation / nucleoplasm / nucleus / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 1.6 Å X-RAY DIFFRACTION / Resolution: 1.6 Å | ||||||
 Authors Authors | Lalonde, J.M. / Bernlohr, D.A. / Banaszak, L.J. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1994 Title: X-ray crystallographic structures of adipocyte lipid-binding protein complexed with palmitate and hexadecanesulfonic acid. Properties of cavity binding sites. Authors: LaLonde, J.M. / Bernlohr, D.A. / Banaszak, L.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1lie.cif.gz | 40 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1lie.ent.gz | 27.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1lie.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/li/1lieftp://data.pdbj.org/pub/pdb/validation_reports/li/1lie | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 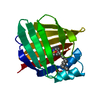
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 14587.687 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
|---|---|
| #2: Chemical | ChemComp-PLM /   Mass: 256.424 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H32O2 Mass: 256.424 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H32O2 |
| #3: Chemical | ChemComp-PPI / 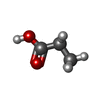  Mass: 74.079 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H6O2 Mass: 74.079 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H6O2 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 82 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 82 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.22 Å3/Da / Density % sol: 44.61 % |
|---|---|
| Crystal grow | *PLUS Method: batch method / PH range low: 7.2 / PH range high: 6.6 |
| Components of the solutions | *PLUS Conc.: 2.1-3.0 M / Chemical formula: NaH2PO4-K2HPO4 |
-Data collection
| Radiation | Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| Reflection | *PLUS Highest resolution: 1.6 Å / Num. obs: 16514 / % possible obs: 96.4 % / Observed criterion σ(I): 2 / Redundancy: 5.3 % / Num. measured all: 73035 / Rmerge(I) obs: 0.0363 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.6→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.6→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.198 / Rfactor Rfree: 0.239 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 15.9 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: x_angle_d / Dev ideal: 1.65 |
