 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1l9w: CRYSTAL STRUCTURE OF 3-DEHYDROQUINASE FROM SALMONELLA TYPHI COMPL... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1l9w | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF 3-DEHYDROQUINASE FROM SALMONELLA TYPHI COMPLEXED WITH REACTION PRODUCT | ||||||
 Components Components | 3-dehydroquinate dehydratase aroD | ||||||
 Keywords Keywords | LYASE / TIM-Barrel / complex with product | ||||||
| Function / homology |  Function and homology information Function and homology information3,4-dihydroxybenzoate biosynthetic process / 3-dehydroquinate dehydratase / 3-dehydroquinate dehydratase activity / chorismate biosynthetic process / aromatic amino acid biosynthetic process / amino acid biosynthetic process Similarity search - Function | ||||||
| Biological species |  Salmonella typhi (bacteria) Salmonella typhi (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.1 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.1 Å | ||||||
 Authors Authors | Lee, W.H. / Perles, L.A. / Nagem, R.A.P. / Shrive, A.K. / Hawkins, A. / Sawyer, L. / Polikarpov, I. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2002 Title: Comparison of different crystal forms of 3-dehydroquinase from Salmonella typhi and its implication for the enzyme activity. Authors: Lee, W.H. / Perles, L.A. / Nagem, R.A. / Shrive, A.K. / Hawkins, A. / Sawyer, L. / Polikarpov, I. | ||||||
| History |
| ||||||
| Remark 600 | HETEROGEN DHS: 3-AMINO-4,5-DIHYDROXY-CYCLOHEX-1-ENECARBOXYLATE GROUP. Formula:4(C7 H8 N1 O4). THE ...HETEROGEN DHS: 3-AMINO-4,5-DIHYDROXY-CYCLOHEX-1-ENECARBOXYLATE GROUP. Formula:4(C7 H8 N1 O4). THE PRODUCT IS COVALENTLY LINKED TO LYS170 THROUGH FORMATION OF A SCHIFF BASE (IMINE) INTERMEDIATE THAT IS THEN REDUCED WITH BOROHYDRIDE. THE EMPIRICAL FORMULA GIVEN CONTAINS THE CARBOXYLATE PROTON BUT NOT THE 3-KETO OXYGEN (WHICH IS REPLACED BY THE NZ OF LYS170 IN FORMING THE SCHIFF BASE). LYS 170 FORMS THE IMINE INTERMEDIATE WITH THE AID OF HIS 143. ARG 213 INTERACTS WITH THE CARBOXYL GROUP OF THE 3-DEHYDROQUINATE SUBSTRATE. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1l9w.cif.gz | 203.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1l9w.ent.gz | 164.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1l9w.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/l9/1l9wftp://data.pdbj.org/pub/pdb/validation_reports/l9/1l9w | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1gqnC  1qfeS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 27654.912 Da / Num. of mol.: 4 / Source method: isolated from a natural source Details: THE PRODUCT 3-DEHYDROSHIKIMATE IS COVALENTLY ATTACHED TO THE ACTIVE SITE LYS 170 BY BOROHYDRIDE REDUCTION OF THE IMINE (SCHIFF BASE) Source: (natural) Salmonella typhi (bacteria) / References: UniProt: P24670, 3-dehydroquinate dehydratase#2: Chemical | ChemComp-DHS / 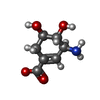  Mass: 172.159 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C7H10NO4 Mass: 172.159 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C7H10NO4#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 207 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 207 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 3 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.62 Å3/Da / Density % sol: 53 % | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 5.5 Details: PEG 4000, citrate-phosphate, pH 5.5, VAPOR DIFFUSION, HANGING DROP, temperature 293.0K | ||||||||||||||||||
| Crystal grow | *PLUS Method: sparse matrix method / PH range low: 6 / PH range high: 5 | ||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 277 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: X31 / Wavelength: 0.928 Å / Beamline: X31 / Wavelength: 0.928 Å |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE |
| Radiation | Monochromator: DOUBLE CRYSTAL / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.928 Å / Relative weight: 1 |
| Reflection | Resolution: 2.1→10 Å / Num. all: 49700 / Num. obs: 49700 / % possible obs: 76.45 % / Observed criterion σ(F): 2 / Observed criterion σ(I): 2 |
| Reflection shell | Resolution: 2.1→2.15 Å / % possible all: 70.12 |
| Reflection | *PLUS Highest resolution: 2.1 Å / Redundancy: 3.42 % / Rmerge(I) obs: 0.069 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1QFE Resolution: 2.1→10 Å / Cross valid method: THROUGHOUT / σ(F): 2
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→10 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.1 Å / % reflection Rfree: 5 % | |||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
