 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1l9l | ||||||
|---|---|---|---|---|---|---|---|
| Title | GRANULYSIN FROM HUMAN CYTOLYTIC T LYMPHOCYTES | ||||||
 Components Components | Granulysin | ||||||
 Keywords Keywords | ANTIMICROBIAL PROTEIN / GRANULYSIN / SAPOSIN FOLD / MEMBRANE-LYTIC | ||||||
| Function / homology |  Function and homology information Function and homology informationphagocytic vesicle lumen / Antimicrobial peptides / defense response to fungus / cellular defense response / antimicrobial humoral immune response mediated by antimicrobial peptide / killing of cells of another organism / defense response to bacterium / : / extracellular region Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 0.92 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 0.92 Å | ||||||
 Authors Authors | Anderson, D.H. / Sawaya, M.R. / Cascio, D. / Ernst, W. / Krensky, A. / Modlin, R. / Eisenberg, D. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2002 Title: Granulysin Crystal Structure and a Structure-Derived Lytic Mechanism Authors: Anderson, D.H. / Sawaya, M.R. / Cascio, D. / Ernst, W. / Modlin, R. / Krensky, A. / Eisenberg, D. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1l9l.cif.gz | 66.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1l9l.ent.gz | 49.6 KB | Display | PDB format |
| PDBx/mmJSON format | 1l9l.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/l9/1l9lftp://data.pdbj.org/pub/pdb/validation_reports/l9/1l9l | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 8679.092 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Cell: LYMPHOCYTE / Plasmid: pET28a (Novagen) / Species (production host): Escherichia coli / Production host:  | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| #2: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4#3: Chemical | ChemComp-MPO / |   Mass: 209.263 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H15NO4S / Comment: pH buffer*YM Mass: 209.263 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H15NO4S / Comment: pH buffer*YM#4: Chemical | 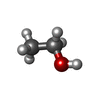  Mass: 46.068 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H6O Mass: 46.068 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H6O#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 93 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 93 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.55 Å3/Da / Density % sol: 20.6 % Description: SE-MET PATTERSON PEAKS WERE 10 SIGMA. PHASING FROM 2 SITES, PLUS INPUT OF SEQUENCE WAS ENOUGH FOR ARP/WARP TO AUTO-BUILD ALMOST THE ENTIRE STRUCTURE. | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 294 K / Method: vapor diffusion, hanging drop / pH: 7 Details: 3.1-3.2 M AMMONIUM SULFATE, 0.05 M N-MORPHOLINO PROPANESULFONIC ACID TITRATED TO PH 7 WITH SODIUM HYDROXIDE, 2.5% ETHANOL., VAPOR DIFFUSION, HANGING DROP, temperature 294K | ||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 21-22 ℃ | ||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X8C / Wavelength: 0.9795 / Beamline: X8C / Wavelength: 0.9795 |
| Detector | Type: ADSC QUANTUM 4r / Detector: CCD / Date: Feb 10, 2002 / Details: PARABOLIC COLLIMATING MIR |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9795 Å / Relative weight: 1 |
| Reflection | Resolution: 0.92→23.6 Å / Num. all: 35664 / Num. obs: 35664 / % possible obs: 82.3 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -3 / Redundancy: 7.1 % / Rmerge(I) obs: 0.037 / Net I/σ(I): 48.3 |
| Reflection shell | Resolution: 0.92→0.96 Å / Redundancy: 3.7 % / Rmerge(I) obs: 0.304 / Mean I/σ(I) obs: 3.8 / Num. unique all: 1368 / % possible all: 31.7 |
| Reflection | *PLUS |
| Reflection shell | *PLUS % possible obs: 31.7 % / Rmerge(I) obs: 0.3 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 0.92→23.59 Å / Num. parameters: 7094 / Num. restraintsaints: 9130 / Cross valid method: FREE R / σ(F): 0 / σ(I): -3 StereochEM target val spec case: SULFATE AND ETHANOL COORDINATES WERE OBTAINED FROM CAMBRIDGE CRYSTAL STRUCTURE DATABASE, AND USED AS TARGETS. N-MORPHOLINO PROPANESULFONATE COORDINATES WERE OBTAINED ...StereochEM target val spec case: SULFATE AND ETHANOL COORDINATES WERE OBTAINED FROM CAMBRIDGE CRYSTAL STRUCTURE DATABASE, AND USED AS TARGETS. N-MORPHOLINO PROPANESULFONATE COORDINATES WERE OBTAINED FROM HIC-UP AND SYMMETRIZED FOR TARGET VALUES Stereochemistry target values: ENGH & HUBER Details: WATER MOLECULES WITH APPARENT COLLISIONS: NOT ALL WATERS IN THE MODEL CAN BE PRESENT SIMULTANEOUSLY. HOH 1021 IN POSITIONS A AND B CORRESPONDS TO ASP 72 IN CONFORMATIONS A AND B, ...Details: WATER MOLECULES WITH APPARENT COLLISIONS: NOT ALL WATERS IN THE MODEL CAN BE PRESENT SIMULTANEOUSLY. HOH 1021 IN POSITIONS A AND B CORRESPONDS TO ASP 72 IN CONFORMATIONS A AND B, RESPECTIVELY. HOH 1041 IN POSITIONS A AND B CORRESPONDS TO GLN 12 IN CONFORMATIONS A AND B, RESPECTIVELY. HOH 1080 IN POSITION B CORRESPONDS TO ASP 43 IN CONFORMATION B; IT WOULD COLLIDE WITH CONFORMATION A. GLN 12 AND ASP 72 OF A SYMMETRY-RELATED MOLECULE CAN SIMULTANEOUSLY OCCUPY THEIR CONFORMATIONS A, ALONG WITH HOH 1021A AND 1041A, BUT EITHER SIDE CHAIN IN ITS CONFORMATION A WILL EXCLUDE HOH 1021B.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: MOEWS & KRETSINGER, J.MOL.BIOL.91(1973)201-228 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 13 / Occupancy sum hydrogen: 620.82 / Occupancy sum non hydrogen: 727.7 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 0.92→23.59 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: SHELXL / Version: 97 / Classification: refinement | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS % reflection Rfree: 5 % / Rfactor Rwork: 0.138 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
