 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1l5h | ||||||
|---|---|---|---|---|---|---|---|
| Title | FeMo-cofactor Deficient Nitrogenase MoFe Protein | ||||||
 Components Components |
| ||||||
 Keywords Keywords | OXIDOREDUCTASE / apo-protein | ||||||
| Function / homology |  Function and homology information Function and homology informationnitrogen fixation / molybdenum-iron nitrogenase complex / nitrogenase / nitrogenase activity / iron-sulfur cluster binding / ATP binding / metal ion binding Similarity search - Function | ||||||
| Biological species |  Azotobacter vinelandii (bacteria) Azotobacter vinelandii (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.3 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.3 Å | ||||||
 Authors Authors | Schmid, B. / Ribbe, M.W. / Einsle, O. / Yoshida, M. / Thomas, L.M. / Dean, D.R. / Rees, D.C. / Burgess, B.K. | ||||||
 Citation Citation | Journal: Science / Year: 2002 Title: Structure of a cofactor-deficient nitrogenase MoFe protein. Authors: Schmid, B. / Ribbe, M.W. / Einsle, O. / Yoshida, M. / Thomas, L.M. / Dean, D.R. / Rees, D.C. / Burgess, B.K. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1l5h.cif.gz | 201.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1l5h.ent.gz | 158.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1l5h.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/l5/1l5hftp://data.pdbj.org/pub/pdb/validation_reports/l5/1l5h | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3minS S: Starting model for refinement |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | The second part of the biological assembly is generated by the two fold axis |
-Components
| #1: Protein | Mass: 55231.848 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Azotobacter vinelandii (bacteria) / References: UniProt: P07328, nitrogenase |
|---|---|
| #2: Protein | Mass: 59404.684 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Azotobacter vinelandii (bacteria) / References: UniProt: P07329, nitrogenase |
| #3: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca |
| #4: Chemical | ChemComp-CLF / 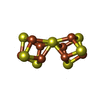  Mass: 671.215 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe8S7 Mass: 671.215 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe8S7 |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 241 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 241 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.22 Å3/Da / Density % sol: 61.81 % | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: liquid diffusion / pH: 9.5 Details: PEG 8000, CHES, pH 9.5, LIQUID DIFFUSION, temperature 298K | ||||||||||||||||||
| Crystal grow | *PLUS Method: batch method | ||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL11-1 / Wavelength: 0.965 Å / Beamline: BL11-1 / Wavelength: 0.965 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: May 9, 2001 |
| Radiation | Monochromator: GRAPHITE / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.965 Å / Relative weight: 1 |
| Reflection | Resolution: 2.3→30 Å / Num. all: 65889 / Num. obs: 60354 / % possible obs: 91.6 % / Observed criterion σ(I): 2 / Rmerge(I) obs: 0.088 / Net I/σ(I): 15.7 |
| Reflection shell | Resolution: 2.3→2.38 Å / Rmerge(I) obs: 0.44 / Mean I/σ(I) obs: 2.2 / % possible all: 87.5 |
| Reflection | *PLUS Num. measured all: 1632866 / Rmerge(I) obs: 0.088 |
| Reflection shell | *PLUS % possible obs: 87.5 % / Rmerge(I) obs: 0.44 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 3MIN Resolution: 2.3→30 Å / Isotropic thermal model: Isotropic / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 2 / Stereochemistry target values: Engh & Huber
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.3→30 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| Refinement | *PLUS % reflection Rfree: 5 % / Rfactor obs: 0.249 / Rfactor Rfree: 0.289 / Rfactor Rwork: 0.249 | |||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
