 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1kmy: Crystal Structure of 2,3-dihydroxybiphenyl 1,2-dioxygenase Comple... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1kmy | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of 2,3-dihydroxybiphenyl 1,2-dioxygenase Complexed with 2,3-dihydroxybiphenyl under Anaerobic Condition | ||||||
 Components Components | 2,3-DIHYDROXYBIPHENYL 1,2-DIOXYGENASE | ||||||
 Keywords Keywords | OXIDOREDUCTASE / dioxygenase / 2 / 3-dihydroxybiphenyl | ||||||
| Function / homology |  Function and homology information Function and homology informationbiphenyl-2,3-diol 1,2-dioxygenase / biphenyl-2,3-diol 1,2-dioxygenase activity / xenobiotic catabolic process / ferrous iron binding Similarity search - Function | ||||||
| Biological species |  Burkholderia xenovorans (bacteria) Burkholderia xenovorans (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Han, S. / Bolin, J.T. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 1998 Title: Molecular basis for the stabilization and inhibition of 2, 3-dihydroxybiphenyl 1,2-dioxygenase by t-butanol. Authors: Vaillancourt, F.H. / Han, S. / Fortin, P.D. / Bolin, J.T. / Eltis, L.D. #1: Journal: TO BE PUBLISHEDTitle: Definitive Evidence for Monoanionic Binding of 2,3-dihydroxybiphenyl to 2,3-dihydroxybiphenyl Dioxygenase from UV Resonance Raman Spectroscopy, UV/Vis Absorption Spectroscopy, and Crystallography. Authors: Vaillancourt, F.H. / Barbosa, C.J. / Spiro, T.G. / Bolin, J.T. / Blades, M.W. / Turner, R.F.B. / Eltis, L.D. #2: Journal: Science / Year: 1995Title: Crystal Structure of the Biphenyl-cleaving Extradiol Dioxygenase from a PCB-degrading Pseudomonad. Authors: Han, S. / Eltis, L.D. / Timmis, K.N. / Muchmore, S.W. / Bolin, J.T. #3: Journal: Handbook of Metalloproteins / Year: 2001Title: 2,3-Dihydroxybiphenyl 1,2-dioxygenase. Authors: Bolin, J.T. / Eltis, L.D. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1kmy.cif.gz | 75.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1kmy.ent.gz | 55.6 KB | Display | PDB format |
| PDBx/mmJSON format | 1kmy.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/km/1kmyftp://data.pdbj.org/pub/pdb/validation_reports/km/1kmy | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1kndC  1knfC  1hanS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | x 8
| ||||||||
| Unit cell |
| ||||||||
| Details | The biological assembly is a homo-octamer generated by crystallographic symmetry |
-Components
| #1: Protein | Mass: 32377.598 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Burkholderia xenovorans (bacteria) / Strain: LB400Description: HYPEREXPRESSED IN THE PARENT STRAIN. (This organism has been reclassified. Prior publications may refer to this source as Pseudomonas sp. strain LB400.) Gene: BPHC / Plasmid: PLEBD4 / Production host: Burkholderia cepacia (bacteria)References: UniProt: P47228, biphenyl-2,3-diol 1,2-dioxygenase |
|---|---|
| #2: Chemical | ChemComp-FE2 /   Mass: 55.845 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe Mass: 55.845 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe |
| #3: Chemical | ChemComp-BPY /   Mass: 186.207 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H10O2 Mass: 186.207 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H10O2 |
| #4: Chemical | ChemComp-TBU / 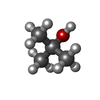  Mass: 74.122 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O Mass: 74.122 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 115 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 115 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.22 Å3/Da / Density % sol: 62 % |
|---|---|
| Crystal grow | Temperature: 278 K / Method: vapor diffusion, sitting drop / pH: 7.5 Details: PEG4000, t-butanol, 2,3-dihydroxybiphenyl, pH 7.5, VAPOR DIFFUSION, SITTING DROP, temperature 278K |
| Crystal grow | *PLUS Method: unknown |
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 Å |
| Detector | Type: RIGAKU RAXIS II / Detector: IMAGE PLATE / Date: Nov 21, 1995 / Details: mirrors |
| Radiation | Monochromator: YALE MIRRORS / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2→30 Å / Num. all: 28856 / Num. obs: 28856 / % possible obs: 99.3 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -3 / Redundancy: 5.7 % / Rmerge(I) obs: 0.063 / Net I/σ(I): 34.7 |
| Reflection shell | Resolution: 2→2.07 Å / Redundancy: 3.3 % / Rmerge(I) obs: 0.229 / Mean I/σ(I) obs: 5 / Num. unique all: 2728 / % possible all: 95.2 |
| Reflection | *PLUS Lowest resolution: 30 Å / Rmerge(I) obs: 0.63 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1HAN Resolution: 2→7 Å / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / Stereochemistry target values: Engh & Huber Details: All Fe-ligand bond distances were harmonically restrained to an equilibrium distance of 2.2 Angstroms using a weak force constant of 10 kcal/(mole x Angstrom-squared). Bond length, bond ...Details: All Fe-ligand bond distances were harmonically restrained to an equilibrium distance of 2.2 Angstroms using a weak force constant of 10 kcal/(mole x Angstrom-squared). Bond length, bond angle, and planarity restraints similar to those used for aromatic side chains were applied to het group BPY. The torsion angle between the rings of BPY was unrestrained.
| ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 21.3 Å2 | ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→7 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.03 Å
| ||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2 Å / Lowest resolution: 7 Å / Num. reflection obs: 26435 / σ(F): 0 / Rfactor obs: 0.161 | ||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 21.3 Å2 | ||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Highest resolution: 2 Å / Rfactor Rfree: 0.25 / Num. reflection Rfree: 62 / Rfactor Rwork: 0.238 |
