 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1j47: 3D Solution NMR Structure of the M9I Mutant of the HMG-Box Domain... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1j47 | ||||||
|---|---|---|---|---|---|---|---|
| Title | 3D Solution NMR Structure of the M9I Mutant of the HMG-Box Domain of the Human Male Sex Determining Factor SRY Complexed to DNA | ||||||
 Components Components |
| ||||||
 Keywords Keywords | TRANSCRIPTION/DNA / MALE SEX DETERMINING FACTOR / SRY / SEX-REVERSAL MUTATION / DNA BENDING MUTANT / DIPOLAR COUPLINGS / MULTIDIMENSIONAL NMR / TRANSCRIPTION-DNA COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationpositive regulation of male gonad development / Transcriptional regulation of testis differentiation / sex differentiation / male sex determination / Deactivation of the beta-catenin transactivating complex / DNA-binding transcription activator activity, RNA polymerase II-specific / DNA-binding transcription factor binding / cell differentiation / DNA-binding transcription factor activity, RNA polymerase II-specific / calmodulin binding ...positive regulation of male gonad development / Transcriptional regulation of testis differentiation / sex differentiation / male sex determination / Deactivation of the beta-catenin transactivating complex / DNA-binding transcription activator activity, RNA polymerase II-specific / DNA-binding transcription factor binding / cell differentiation / DNA-binding transcription factor activity, RNA polymerase II-specific / calmodulin binding / nuclear speck / RNA polymerase II cis-regulatory region sequence-specific DNA binding / positive regulation of gene expression / chromatin / positive regulation of DNA-templated transcription / positive regulation of transcription by RNA polymerase II / DNA binding / nucleoplasm / nucleus / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | SOLUTION NMR | ||||||
 Authors Authors | Clore, G.M. / Murphy, E.C. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2001 Title: Structural basis for SRY-dependent 46-X,Y sex reversal: modulation of DNA bending by a naturally occurring point mutation. Authors: Murphy, E.C. / Zhurkin, V.B. / Louis, J.M. / Cornilescu, G. / Clore, G.M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1j47.cif.gz | 66.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1j47.ent.gz | 47.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1j47.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 1j47_validation.pdf.gz | 248.9 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 1j47_full_validation.pdf.gz | 248.6 KB | Display | |
| Data in XML | 1j47_validation.xml.gz | 3.6 KB | Display | |
| Data in CIF | 1j47_validation.cif.gz | 4.8 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/j4/1j47ftp://data.pdbj.org/pub/pdb/validation_reports/j4/1j47 | HTTPS FTP |
-Related structure data
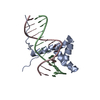







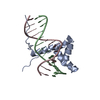
-Links
 PDBj
PDBj






































- Assembly
Assembly
| Deposited unit | 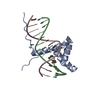
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| NMR ensembles |
|
-Components
| #1: DNA chain | Mass: 4178.747 Da / Num. of mol.: 1 / Source method: obtained synthetically |
|---|---|
| #2: DNA chain | Mass: 4382.835 Da / Num. of mol.: 1 / Source method: obtained synthetically |
| #3: Protein | Mass: 10636.532 Da / Num. of mol.: 1 / Fragment: HMG-BOX DOMAIN / Mutation: M9I Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Species (production host): Escherichia coli / Production host:  |
-Experimental details
-Experiment
| Experiment | Method: SOLUTION NMR |
|---|---|
| NMR details | Text: THE FOLLOWING EXPERIMENTS WERE CONDUCTED: (1) DOUBLE AND TRIPLE RESONANCE FOR ASSIGNMENT OF PROTEIN; (2) DOUBLE RESONANCE AND HETERONUCLEAR FILTERED FOR DNA; (3) QUANTITATIVE J CORRELATION FOR ...Text: THE FOLLOWING EXPERIMENTS WERE CONDUCTED: (1) DOUBLE AND TRIPLE RESONANCE FOR ASSIGNMENT OF PROTEIN; (2) DOUBLE RESONANCE AND HETERONUCLEAR FILTERED FOR DNA; (3) QUANTITATIVE J CORRELATION FOR COUPLING CONSTANTS; (4) 2D, 3D AND 4D HETERONUCLEAR SEPARATED AND FILTERED NOE EXPERIMENTS; (4) 2D and 3D DOUBLE AND TRIPLE RESONANCE EXPERIMENTS FOR DIPOLAR COUPLING MEASUREMENTS IN LIQUID CRYSTALLINE MEDIUM OF 4.5-5% 3:1 DMPC:DHPC |
- Sample preparation
Sample preparation
| Sample conditions | Ionic strength: 50 mM SODIUM PHOSPHATE / pH: 7.2 / Temperature: 308.00 K |
|---|
-NMR measurement
| NMR spectrometer |
|
|---|
- Processing
Processing
| NMR software | Name: X-PLOR NIH VERSION (HTTP://NMR.CIT.NIH.GOV) / Version: (HTTP://NMR.CIT.NIH.GOV) / Developer: CLORE, KUSZEWSKI, SCHWIETERS / Classification: refinement |
|---|---|
| Refinement | Software ordinal: 1 Details: THE STRUCTURE WAS CALCULATED BY SIMULATED ANNEALING IN TORSION ANGLE SPACE (C. SCHWIETERS AND G.M. CLORE. J. MAGN. RESON., IN PRESS). THE TARGET FUNCTION COMPRISES TERMS FOR NOE RESTRAINTS, ...Details: THE STRUCTURE WAS CALCULATED BY SIMULATED ANNEALING IN TORSION ANGLE SPACE (C. SCHWIETERS AND G.M. CLORE. J. MAGN. RESON., IN PRESS). THE TARGET FUNCTION COMPRISES TERMS FOR NOE RESTRAINTS, TORSION ANGLE RESTRAINTS, CARBON CHEMICAL SHIFT RESTRAINTS (KUSZWESKI ET AL. J. MAGN. RESON. SERIES B 106, 92-96 (1995), J COUPLING RESTRAINTS (GARRETT ET AL. J. MAGN. RESON. SERIES B 104, 99, 103 (1994); DIPOLAR COUPLING RESTRAINTS (CLORE ET AL. J.MAGN.RESON. 131, 159-162 (1998); J.MAGN.RESON. 133, 216-221(1998)), AND RADIUS OF GYRATION (KUSZEWSKI ET AL. JACS 121, 2337 (1999)). THE NON-BONDED CONTACTS ARE REPRESENTED BY A QUARTIC VAN DER WAALS REPULSION TERM (NILGES ET AL. (1988) FEBS LETT. 229, 129-136); THE DELPHIC TORSION ANGLE DATABASE POTENTIAL (KUSZEWSKI & C J. MAGN. RESON. 146, 249 (2000)); AND THE DELPHIC BASE-BASE POSITIONAL DATABASE POTENTIAL (KUSZEWSKI ET AL. JACS 123, 3903 (2001)). IN THIS ENTRY THE SECOND TO LAST COLUMN REPRESENTS THE AVERAGE RMS DIFFERENCE BETWEEN THE INDIVIDUAL SIMULATED ANNEALING STRUCTURES (400 FOR THE WILD TYPE COMPLEX) AND THE MEAN COORDINATE POSITIONS (OBTAINED BY BEST FITTING TO RESIDUES 4-81 OF THE PROTEIN AND 101-128 OF THE DNA). THE ORIENTATION PROVIDED FOR THE MUTANT (1J47) RELATIVE TO THE WILD TYPE (1J46) WAS OBTAINED BY BEST-FITTING TO RESIDUES 4-81 OF THE PROTEIN. WILD TYPE M9I MUTANT PDB ID: 1J46 1J47 DEVIATIONS FROM IDEALIZED GEOMETRY: BONDS 0.003 A 0.003 A ANGLES 0.81 DEG 0.80 DEG IMPROPERS 0.79 DEG 0.79 DEG DEVIATIONS FROM EXPT RESTRAINTS NOES (1795/1693) 0.04 A 0.03 A TORSION ANGLES (433/429) 0.29 DEG 0.30 DEG 3JHNA COUPLINGS (70/66) 0.84 HZ 0.90 HZ 13C CHEMICAL SHIFTS (165/165) 0.99 PPM 0.95 PPM HETERONUCLEAR DIPOLAR COUPLING R-FACTORS (CLORE AND GARRETT J. AM. CHEM. SOC. 121, 9008-9012): PROTEIN 1DNH (71/66) 5.5% 7.6% PROTEIN 1DCH (67/67) 6.3% 10.0% PROTEIN 1DNC' (68/62) 18.9% 28.9% PROTEIN 2DHNC'(68/62) 18.8% 21.6% DNA 1DNH (9/10) 10.2% 16.1% DNA 1DCH (37/33) 11.2% 10.7% DNA 1H-1H DIPOLAR COUPLINGS (55/53) 0.56 HZ 0.75 HZ % RESIDUES IN MOST FAVORABLE REGION OF RAMACHADRAN MAP 94.7% 94.7% |
| NMR ensemble | Conformer selection criteria: RESTRAINED REGULARIZED MEAN STRUCTURE Conformers calculated total number: 400 / Conformers submitted total number: 1 |
