 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1ixj: Crystal Structure of d(GCGAAAGCT) Containing Parallel-stranded Du... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ixj | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of d(GCGAAAGCT) Containing Parallel-stranded Duplex with Homo Base Pairs and Anti-Parallel Duplex with Watson-Crick Base pairs | ||||||||||||||||||||
 Components Components | 5'-D(* Keywords KeywordsDNA / Parallel DNA / homo base pairs / parallel-stranded helix / parallel duplex | Function / homology | COBALT HEXAMMINE(III) / DNA |  Function and homology information Function and homology informationMethod |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.5 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.5 Å  Authors AuthorsSunami, T. / Kondo, J. / Kobuna, T. / Hirao, I. / Watanabe, K. / Miura, K. / Takenaka, A. |  CitationJournal: Nucleic Acids Res. / Year: 2002 CitationJournal: Nucleic Acids Res. / Year: 2002Title: Crystal Structure of d(GCGAAAGCT) Containing a Parallel-stranded Duplex with Homo Base Pairs and an Anti-Parallel Duplex with Watson-Crick Base pairs Authors: Sunami, T. / Kondo, J. / Kobuna, T. / Hirao, I. / Watanabe, K. / Miura, K. / Takenaka, A. History |
Remark 300 | BIOMOLECULE: THIS ENTRY CONTAINS THE CRYSTALLOGRAPHIC ASYMMETRIC UNIT WHICH CONSISTS OF 1 CHAIN(S). ... BIOMOLECULE: THIS ENTRY CONTAINS THE CRYSTALLOGRAPHIC ASYMMETRIC UNIT WHICH CONSISTS OF 1 CHAIN(S). BASES 2-5 FORM A PARALLEL STRANDED DUPLEX WITH HOMO BASE PAIRS WITH BASES 2-5 OF THE STRAND GENERATED BY SYMMETRY OPERATOR 8_666 : 1-Y, 1-X, 1-Z. BASES 6-9 FORM AN ANTIPARALLEL DUPLEX WITH WATSON-CRICK BASE PAIRS WITH BASES 9-6 OF THE STRAND GENERATED BY SYMMETRY OPERATOR 5_655 : 3/2-X,Y,3/4-Z | |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ixj.cif.gz | 15.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ixj.ent.gz | 9.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1ixj.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ix/1ixjftp://data.pdbj.org/pub/pdb/validation_reports/ix/1ixj | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|




-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
| #1: DNA chain | Mass: 2764.837 Da / Num. of mol.: 1 / Source method: obtained synthetically | ||||
|---|---|---|---|---|---|
| #2: Chemical |   Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg#3: Chemical | ChemComp-NCO / | 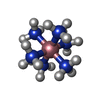  Mass: 161.116 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CoH18N6 Mass: 161.116 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CoH18N6#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 19 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 19 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 2 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.33 Å3/Da / Density % sol: 63.03 % | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 6 Details: 2,6-diaminopyridine, magnesium chloride, sodium chloride, hexaamminecobalt(III) chloride, 2-methyl-2,4-pentanediol, MEGA-10, sodium cacodylate, VAPOR DIFFUSION, HANGING DROP, temperature 293K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 6 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction |
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source |
| |||||||||||||||
| Detector |
| |||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||
| Radiation wavelength |
| |||||||||||||||
| Reflection | Resolution: 2.4→37.9 Å / Num. all: 1689 / Num. obs: 1676 / % possible obs: 100 % / Observed criterion σ(I): 3 / Rmerge(I) obs: 0.068 | |||||||||||||||
| Reflection shell | Resolution: 2.4→2.52 Å / Rmerge(I) obs: 0.239 / Mean I/σ(I) obs: 2.7 / % possible all: 100 | |||||||||||||||
| Reflection | *PLUS Lowest resolution: 27 Å / Num. obs: 1647 / % possible obs: 98.3 % / Num. measured all: 20224 | |||||||||||||||
| Reflection shell | *PLUS Lowest resolution: 2.53 Å / % possible obs: 100 % / Rmerge(I) obs: 0.054 |

- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 2.5→9 Å / σ(F): 3 Stereochemistry target values: G. PARKINSON ET AL., (1996) ACTACRYST. D52, 57-64 Details: The C1*-C2*-C3* angle of the G7 residue is slightly small due to large temperature factor of the C8 residue.
| ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.5→9 Å
| ||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||
| LS refinement shell | Resolution: 2.5→2.61 Å /
| ||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 9 Å / % reflection Rfree: 10 % | ||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||
| Refine LS restraints | *PLUS
|
