 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1it6: CRYSTAL STRUCTURE OF THE COMPLEX BETWEEN CALYCULIN A AND THE CATA... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1it6 | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF THE COMPLEX BETWEEN CALYCULIN A AND THE CATALYTIC SUBUNIT OF PROTEIN PHOSPHATASE 1 | ||||||
 Components Components | SERINE/THREONINE PROTEIN PHOSPHATASE 1 GAMMA (PP1-GAMMA) CATALYTIC SUBUNIT | ||||||
 Keywords Keywords | HYDROLASE / HYDROLASE-INHIBITOR COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationPTW/PP1 phosphatase complex / regulation of nucleocytoplasmic transport / protein phosphatase 1 binding / lamin binding / SHOC2 M1731 mutant abolishes MRAS complex function / Gain-of-function MRAS complexes activate RAF signaling / protein dephosphorylation / Phosphorylation and nuclear translocation of the CRY:PER:kinase complex / glycogen metabolic process / Triglyceride catabolism ...PTW/PP1 phosphatase complex / regulation of nucleocytoplasmic transport / protein phosphatase 1 binding / lamin binding / SHOC2 M1731 mutant abolishes MRAS complex function / Gain-of-function MRAS complexes activate RAF signaling / protein dephosphorylation / Phosphorylation and nuclear translocation of the CRY:PER:kinase complex / glycogen metabolic process / Triglyceride catabolism / protein-serine/threonine phosphatase / entrainment of circadian clock by photoperiod / protein serine/threonine phosphatase activity / cleavage furrow / phosphatase activity / Maturation of hRSV A proteins / microtubule organizing center / mitotic sister chromatid segregation / positive regulation of glial cell proliferation / blastocyst development / phosphoprotein phosphatase activity / Amplification of signal from unattached kinetochores via a MAD2 inhibitory signal / Mitotic Prometaphase / EML4 and NUDC in mitotic spindle formation / Resolution of Sister Chromatid Cohesion / Downregulation of TGF-beta receptor signaling / circadian regulation of gene expression / RAF activation / RHO GTPases Activate Formins / regulation of circadian rhythm / kinetochore / neuron differentiation / Separation of Sister Chromatids / MAPK cascade / presynapse / midbody / spermatogenesis / dendritic spine / mitochondrial outer membrane / nuclear speck / protein domain specific binding / cell division / focal adhesion / protein kinase binding / protein-containing complex binding / nucleolus / glutamatergic synapse / protein-containing complex / mitochondrion / RNA binding / metal ion binding / nucleus / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Kita, A. / Matsunaga, S. / Takai, A. / Kataiwa, H. / Wakimoto, T. / Fusetani, N. / Isobe, M. / Miki, K. | ||||||
 Citation Citation | Journal: Structure / Year: 2002 Title: Crystal structure of the complex between calyculin A and the catalytic subunit of protein phosphatase 1. Authors: Kita, A. / Matsunaga, S. / Takai, A. / Kataiwa, H. / Wakimoto, T. / Fusetani, N. / Isobe, M. / Miki, K. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1it6.cif.gz | 134 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1it6.ent.gz | 103.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1it6.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/it/1it6ftp://data.pdbj.org/pub/pdb/validation_reports/it/1it6 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1fjmS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 37030.777 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Production host:  References: UniProt: P36873, protein-serine/threonine phosphatase #2: Chemical | ChemComp-MN /   Mass: 54.938 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mn Mass: 54.938 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mn#3: Chemical | 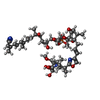  Mass: 1009.170 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C50H81N4O15P Mass: 1009.170 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C50H81N4O15P#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 96 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 96 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.78 Å3/Da / Density % sol: 55.74 % | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 7.8 Details: PEG3350, POTASSIUM SULFATE, MANGANESE(II) CHLORIDE, pH 7.8, VAPOR DIFFUSION, SITTING DROP, temperature 293K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 20 ℃ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 293 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SPring-8  / Beamline: BL44B2 / Wavelength: 1 Å / Beamline: BL44B2 / Wavelength: 1 Å |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Mar 14, 2001 |
| Radiation | Monochromator: DOUBLE CRYSTAL MONOCHROMATOR / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2→50 Å / Num. all: 56617 / Num. obs: 56530 / % possible obs: 99.9 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Rmerge(I) obs: 0.078 / Net I/σ(I): 17.1 |
| Reflection shell | Resolution: 2→2.07 Å / Rmerge(I) obs: 0.39 / Mean I/σ(I) obs: 2.2 / % possible all: 99.6 |
| Reflection | *PLUS Lowest resolution: 50 Å / Redundancy: 6.2 % / Num. measured all: 536140 / Rmerge(I) obs: 0.078 |
| Reflection shell | *PLUS Highest resolution: 2 Å / % possible obs: 99.6 % / Rmerge(I) obs: 0.39 |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1FJM Resolution: 2→20 Å / Isotropic thermal model: ISOTROPIC / σ(F): 2 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||
| Displacement parameters | Biso mean: 21.05 Å2 | ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→20 Å
| ||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.182 / Rfactor Rfree: 0.218 / Rfactor Rwork: 0.182 | ||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||
| Refine LS restraints | *PLUS
|
