 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 1ijw | ||||||
|---|---|---|---|---|---|---|---|
| タイトル | Testing the Water-Mediated Hin Recombinase DNA Recognition by Systematic Mutations. | ||||||
 要素 要素 |
| ||||||
 キーワード キーワード | DNA BINDING PROTEIN/DNA / water-mediated recognition / protein-DNA complex / Hin recombinase / BR18 / DNA BINDING PROTEIN-DNA COMPLEX | ||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報 | ||||||
| 手法 |  X線回折 / シンクロトロン / 多重同系置換・異常分散 / 解像度: 2.4 Å X線回折 / シンクロトロン / 多重同系置換・異常分散 / 解像度: 2.4 Å | ||||||
 データ登録者 データ登録者 | Chiu, T.K. / Sohn, C. / Johnson, R.C. / Dickerson, R.E. | ||||||
 引用 引用 | ジャーナル: EMBO J. / 年: 2002 タイトル: Testing water-mediated DNA recognition by the Hin recombinase. 著者: Chiu, T.K. / Sohn, C. / Dickerson, R.E. / Johnson, R.C. #1: ジャーナル: Thesis / 年: 2001タイトル: How Hin Recombinase, FIS and Cations Bind DNA. Chapter 4. Water-Mediated Sequence-Specific Recognition by Hin Recombinase. 著者: Chiu, T.K. #2: ジャーナル: Science / 年: 1994タイトル: Hin Recombinase Bound to DNA: the Origin of Specificity in Major and Minor Groove Interactions. 著者: Feng, J.A. / Johnson, R.C. / Dickerson, R.E. | ||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 1ijw.cif.gz | 39.7 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb1ijw.ent.gz | 25.1 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 1ijw.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| 文書・要旨 | 1ijw_validation.pdf.gz | 457.9 KB | 表示 | wwPDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | 1ijw_full_validation.pdf.gz | 460 KB | 表示 | |
| XML形式データ | 1ijw_validation.xml.gz | 6 KB | 表示 | |
| CIF形式データ | 1ijw_validation.cif.gz | 7.3 KB | 表示 | |
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/ij/1ijwftp://data.pdbj.org/pub/pdb/validation_reports/ij/1ijw | HTTPS FTP |
-関連構造データ







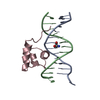
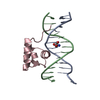



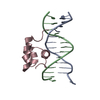


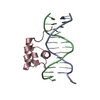

-リンク
 PDBj
PDBj






































- 集合体
集合体
| 登録構造単位 | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| 単位格子 |
|
-要素
| #1: DNA鎖 | 分子量: 4324.836 Da / 分子数: 1 / 由来タイプ: 合成 | ||
|---|---|---|---|
| #2: DNA鎖 | 分子量: 4310.702 Da / 分子数: 1 / 由来タイプ: 合成 | ||
| #3: タンパク質 | 分子量: 6047.051 Da / 分子数: 1 / Fragment: RESIDUES 139 TO 190 / 由来タイプ: 合成 / 詳細: SYNTHETIC PEPTIDE / 参照: UniProt: P03013 | ||
| #4: 化合物 |   分子量: 122.143 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C4H12NO3 / コメント: pH緩衝剤*YM 分子量: 122.143 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C4H12NO3 / コメント: pH緩衝剤*YM#5: 水 | ChemComp-HOH / |  分子量: 18.015 Da / 分子数: 7 / 由来タイプ: 天然 / 式: H2O 分子量: 18.015 Da / 分子数: 7 / 由来タイプ: 天然 / 式: H2O |
-実験情報
-実験
| 実験 | 手法: X線回折 / 使用した結晶の数: 1 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 2.66 Å3/Da / 溶媒含有率: 52 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 結晶化 | 温度: 281 K / 手法: 蒸気拡散法, ハンギングドロップ法 / pH: 8.5 詳細: Hanging drop vapor diffusion at 4C, with initial concentration in drop of 0.13 mM DNA, 0.33 mM Hin, 46 mMTris (pH 8.5), 46 mM CaCl2, 63 mM NaCl, 11.6% v/v PEG400, and 2.03 mM Na cacodylate. ...詳細: Hanging drop vapor diffusion at 4C, with initial concentration in drop of 0.13 mM DNA, 0.33 mM Hin, 46 mMTris (pH 8.5), 46 mM CaCl2, 63 mM NaCl, 11.6% v/v PEG400, and 2.03 mM Na cacodylate. Reservoir solution contains 100 mM Tris (pH 8.5), 100 mM CaCl2, 100 mM NaCl, and 25% PEG400. Concentration of PEG400 in reservoir solution was increased in 5% increments to 35%., VAPOR DIFFUSION, HANGING DROP, temperature 281K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶液の組成 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 結晶化 | *PLUS 温度: 4 ℃ / 手法: 蒸気拡散法, シッティングドロップ法 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶液の組成 | *PLUS
|
-データ収集
| 回折 | 平均測定温度: 100 K 詳細: MULTIPLE WAVELENGTH WITH ANOMALOUS SCATTERING USING THREE IODINE AND ONE BROMINE DERIVATIVES. THE IODINE DERIVATIVES REPLACE THYMINE AT POSITIONS 4, 5, AND 7+19 WITH 5-IODO URACIL. THE ...詳細: MULTIPLE WAVELENGTH WITH ANOMALOUS SCATTERING USING THREE IODINE AND ONE BROMINE DERIVATIVES. THE IODINE DERIVATIVES REPLACE THYMINE AT POSITIONS 4, 5, AND 7+19 WITH 5-IODO URACIL. THE BROMINE DERIVATIVE REPLACES CYTOSINE AT POSITION 18 WITH 5-BROMO CYTOSINE. |
|---|---|
| 放射光源 | 由来: シンクロトロン / サイト: NSLS  / ビームライン: X12C / 波長: 0.917 / ビームライン: X12C / 波長: 0.917 |
| 検出器 | 検出器: CCD / 日付: 1999年6月1日 |
| 放射 | プロトコル: MAD / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray |
| 放射波長 | 波長: 0.917 Å / 相対比: 1 |
| 反射 | 解像度: 2.4→30 Å / Num. obs: 5435 / % possible obs: 85.35 % / Observed criterion σ(I): 0 / 冗長度: 26 % / Biso Wilson estimate: 47 Å2 / Rsym value: 0.079 / Net I/σ(I): 16.59 |
| 反射 シェル | 解像度: 2.4→2.51 Å / 冗長度: 1.91 % / Mean I/σ(I) obs: 2.6 / Rsym value: 0.27 / % possible all: 43.76 |
| 反射 | *PLUS % possible obs: 85.4 % / 冗長度: 26 % / Rmerge(I) obs: 0.079 |
| 反射 シェル | *PLUS % possible obs: 43.8 % / Rmerge(I) obs: 0.27 / Mean I/σ(I) obs: 2.6 |
- 解析
解析
| ソフトウェア |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 構造決定の手法: 多重同系置換・異常分散 開始モデル: MIRAS PHASES 解像度: 2.4→30 Å / Data cutoff high rms absF: 10000 / Isotropic thermal model: ANISOTROPIC_FIXED_ISOTROPIC / σ(F): 0 / 立体化学のターゲット値: MLHL
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶媒の処理 | Bsol: 50 Å2 / ksol: 0.3 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | Biso mean: 40 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化ステップ | サイクル: LAST / 解像度: 2.4→30 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS精密化 シェル | 解像度: 2.4→2.51 Å / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ソフトウェア | *PLUS 名称: CNS / 分類: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化 | *PLUS % reflection Rfree: 10 % / Rfactor Rfree: 0.279 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶媒の処理 | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | *PLUS Biso mean: 40 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS精密化 シェル | *PLUS Rfactor Rfree: 0.421 / % reflection Rfree: 5.1 % / Rfactor Rwork: 0.424 / Rfactor obs: 0.424 |
