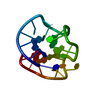
Journal: J.Mol.Biol. / Year: 2001 Title: A double chain reversal loop and two diagonal loops define the architecture of a unimolecular DNA quadruplex containing a pair of stacked G(syn)-G(syn)-G(anti)-G(anti) tetrads flanked by a G- ...Title: A double chain reversal loop and two diagonal loops define the architecture of a unimolecular DNA quadruplex containing a pair of stacked G(syn)-G(syn)-G(anti)-G(anti) tetrads flanked by a G-(T-T) Triad and a T-T-T triple. Authors: Kuryavyi, V. / Majumdar, A. / Shallop, A. / Chernichenko, N. / Skripkin, E. / Jones, R. / Patel, D.J.
STRUCTURES WITH ACCEPTABLE COVALENT GEOMETRY, STRUCTURES WITH FAVORABLE NON-BOND ENERGY,STRUCTURES WITH THE LEAST RESTRAINT VIOLATIONS,BACK-CALCULATED DATA AGREE WITH EXPERIMENTAL NOESY SPECTRUM
Mass: 6258.019 Da / Num. of mol.: 1 / Source method: obtained synthetically
-
Experimental details
-
Experiment
Experiment
Method: SOLUTION NMR
NMR experiment
Conditions-ID
Experiment-ID
Solution-ID
Type
1
1
1
NOESY
1
2
1
TOCSY
1
3
1
DQF-COSY
1
4
1
COSY-45
1
5
1
1
6
1
1
7
1
1
8
1
1
9
1
1
10
1
NMR details
Text: SITE-SPECIFIC SUBSTITUTED SAMPLES CONTAINING 8BR-A,8BR-G,DU,5BR-DU, 2,6A, I, 5ME-C ANALOGS AND SITE-SPECIFIC OR UNIFORMLY 13C AND 15N LABELED SAMPLES HAVE BEEN USED
-
Sample preparation
Details
Solution-ID
Contents
Solvent system
1
5-10 MM DNA (SINGLE STRANDS); 5MM SODIUM PHOSPHATE BUFFER
90% H2O/10% D2O
2
5-10MM DNA (SINGLE STRANDS); 5MM SODIUM PHOSPHATE BUFFER
Method: DISTANCE GEOMETRY, SIMULATED ANNEALING, DISTANCE RESTRAINED MOLECULAR DYNAMICS REFINEMENT, RELAXATION MATRIX INTENSITY REFINEMENT Software ordinal: 1 Details: A TOTAL OF 100 INITIAL DNA STRUCTURES WERE GENERATED USING THE METRIC MATRIX DISTANCE GEOMETRY PROTOCOL OF X-PLOR WITH THE DISTANCE RESTRAINTS OBTAINED EXPERIMENTALLY SPECIFIED AS NOE. ...Details: A TOTAL OF 100 INITIAL DNA STRUCTURES WERE GENERATED USING THE METRIC MATRIX DISTANCE GEOMETRY PROTOCOL OF X-PLOR WITH THE DISTANCE RESTRAINTS OBTAINED EXPERIMENTALLY SPECIFIED AS NOE. FOURTEEN STRUCTURES WERE IDENTIFIED BASED ON THE CRITERION ACCEPTABLE COVALENT GEOMETRY, LOW DISTANCE RESTRAINT VIOLATIONS AND FAVORABLE NON-BONDED ENERGY. THE VARIATION IN ENERGIES WITHIN THIS SUBSET OF STRUCTURES WAS 46 KCAL/MOL AND WAS SEPARATED FROM THE REMAINING STRUCTURES BY A GAP OF 200 KCAL/MOL. THE TEN BEST STRUCTURES FROM THIS SUBSET OF FOURTEEN WERE FURTHER OPTIMIZED WITH RESTRAINED MOLECULAR DYNAMICS, MOLECULAR MECHANICS AND RELAXATION MATRIX INTENSITY REFINEMENT. THE PROTOCOLS ARE AS FOLLOWS. THE DYNAMICS WAS INITIATED AT 300K AND THE TEMPERATURE WAS INCREASED TO 1000K DURING 7PS. AFTER 0.5 PS THE FORCE CONSTANTS WERE GRADUALLY SCALED DURING 17.5 PSEC IN THE PROPORTION 1: 4: 8 FOR THREE CLASSES OF NOE: EXCHANGEABLE, NONEXCHANGEABLE AND HYDROGEN BONDS, RESP. THE STRUCTURES WERE THEN SLOWLY COOLED TO 300K IN 2.8 PS AND EQUILIBRATED AT 300K FOR 12 PS. SOFT PLANARITY RESTRAINTS WERE SWITCHED OFF BEFORE THE EQUILIBRATION STAGE.THE COORDINATES SAVED EVERY 0.5 PS DURING THE LAST 4.0 PS WERE AVERAGED AND THE AVERAGE STRUCTURE WAS SUBJECTED TO 3000 STEPS OF MINIMIZATION. THE DIHEDRAL ANGLE RESTRAINTS AND HYDROGEN BONDS OF THE GGGG TETRADS WERE MAINTAINED THROUGHOUT. THE OBTAINED STRUCTURES WERE SUBJECTED TO RELAXATION MATRIX INTENSITY REFINEMENT. THE INTENSITY VOLUMES WERE USED AS RESTRAINTS AND NON-EXCHANGEABLE PROTON DISTANCE RESTRAINTS WITH THE SCALE FACTOR OF 0.1 RELATIVE TO THE INTENSITY RESTRAINTS. SOFT PLANARITY RESTRAINTS MAINTAINED DURING RELAXATION REFINEMENTS WERE SWITCHED OFF BEFORE THE FINAL STEPS OF MINIMIZATION. THE RELAXATION PATHWAYS WERE CALCULATED USING CUTOFF 4.5 ANGSTROM AND 7.91 NS ISOTROPIC CORRELATION TIME. THE STRUCTURES EXHIBIT PAIRWISE R.M.S.D. VALUE OF 1.07 A.
NMR representative
Selection criteria: closest to the average, fewest violations
NMR ensemble
Conformer selection criteria: STRUCTURES WITH ACCEPTABLE COVALENT GEOMETRY, STRUCTURES WITH FAVORABLE NON-BOND ENERGY,STRUCTURES WITH THE LEAST RESTRAINT VIOLATIONS,BACK-CALCULATED DATA AGREE WITH ...Conformer selection criteria: STRUCTURES WITH ACCEPTABLE COVALENT GEOMETRY, STRUCTURES WITH FAVORABLE NON-BOND ENERGY,STRUCTURES WITH THE LEAST RESTRAINT VIOLATIONS,BACK-CALCULATED DATA AGREE WITH EXPERIMENTAL NOESY SPECTRUM Conformers calculated total number: 100 / Conformers submitted total number: 10
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads






 PDBj
PDBj






































 Assembly
Assembly
 Sample preparation
Sample preparation Processing
Processing X-PLOR
X-PLOR