 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1huv: CRYSTAL STRUCTURE OF A SOLUBLE MUTANT OF THE MEMBRANE-ASSOCIATED ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1huv | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF A SOLUBLE MUTANT OF THE MEMBRANE-ASSOCIATED (S)-MANDELATE DEHYDROGENASE FROM PSEUDOMONAS PUTIDA AT 2.15A RESOLUTION | ||||||
 Components Components | L(+)-MANDELATE DEHYDROGENASE | ||||||
 Keywords Keywords | OXIDOREDUCTASE / TIM BARREL | ||||||
| Function / homology |  Function and homology information Function and homology information(S)-mandelate dehydrogenase / (S)-mandelate dehydrogenase activity / oxidative photosynthetic carbon pathway / (S)-2-hydroxy-acid oxidase / (S)-2-hydroxy-acid oxidase activity / (R)-mandelate catabolic process / L-lactate dehydrogenase (NAD+) activity / hydrogen peroxide biosynthetic process / aerobic respiration / FMN binding ...(S)-mandelate dehydrogenase / (S)-mandelate dehydrogenase activity / oxidative photosynthetic carbon pathway / (S)-2-hydroxy-acid oxidase / (S)-2-hydroxy-acid oxidase activity / (R)-mandelate catabolic process / L-lactate dehydrogenase (NAD+) activity / hydrogen peroxide biosynthetic process / aerobic respiration / FMN binding / peroxisome / plasma membrane Similarity search - Function | ||||||
| Biological species |  Pseudomonas putida (bacteria) Pseudomonas putida (bacteria) Spinacia oleracea (spinach) Spinacia oleracea (spinach) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT, SIR / Resolution: 2.15 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT, SIR / Resolution: 2.15 Å | ||||||
 Authors Authors | Mathews, F.S. / Sukumar, N. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2001 Title: Structure of an active soluble mutant of the membrane-associated (S)-mandelate dehydrogenase. Authors: Sukumar, N. / Xu, Y. / Gatti, D.L. / Mitra, B. / Mathews, F.S. | ||||||
| History |
| ||||||
| Remark 999 | Sequence chimeric mutant of (s)-mandelate dehydrogenase with residues 177-215 replaced by residues ...Sequence chimeric mutant of (s)-mandelate dehydrogenase with residues 177-215 replaced by residues 176-195 of glycolate oxidase. The authors believe SwissProt entry P20932 incorrectly lists residue 15 as ARG instead of ALA. Sequencing of MDH, as well as the original clone pSCR4, has been done a number of times including site-specific mutants and repeatedly confirmed that it is ALA 15 rather than ARG 15. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1huv.cif.gz | 91.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1huv.ent.gz | 67.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1huv.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hu/1huvftp://data.pdbj.org/pub/pdb/validation_reports/hu/1huv | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1goxS S: Starting model for refinement |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | The biological assembly is a tetramer generated from the monomer in the asymmetric unit by crystallographic 4-fold axes. |
-Components
| #1: Protein | Mass: 42132.273 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Details: 20 RESIDUE INSERTION FROM GLYCOLATE OXIDASE AT RESIDUE 177 Source: (gene. exp.) Pseudomonas putida (bacteria), (gene. exp.) Spinacia oleracea (spinach)Production host: |
|---|---|
| #2: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 |
| #3: Chemical | ChemComp-FMN / 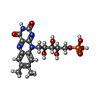  Mass: 456.344 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H21N4O9P Mass: 456.344 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H21N4O9P |
| #4: Chemical | ChemComp-MES /   Mass: 195.237 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H13NO4S / Comment: pH buffer*YM Mass: 195.237 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H13NO4S / Comment: pH buffer*YM |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 291 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 291 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.57 Å3/Da / Density % sol: 52.15 % | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, hanging drop / pH: 6.2 Details: MES, ammonium sulfate, ethylene glycerol, FMN, NaCl, pH 6.2, VAPOR DIFFUSION, HANGING DROP, temperature 291K | |||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
|---|---|
| Detector | Type: RIGAKU RAXIS IV / Detector: IMAGE PLATE |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.15→30 Å / Num. all: 28882 / Num. obs: 28143 / % possible obs: 98.4 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Biso Wilson estimate: 12.7 Å2 / Rsym value: 9.2 / Net I/σ(I): 21.7 |
| Reflection shell | Resolution: 2.15→30 Å / Mean I/σ(I) obs: 6.3 / Rsym value: 36.2 / % possible all: 100 |
| Reflection | *PLUS Rmerge(I) obs: 0.092 |
| Reflection shell | *PLUS % possible obs: 100 % / Rmerge(I) obs: 0.362 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT, SIR Starting model: PDB ENTRY 1GOX Resolution: 2.15→27.42 Å / Rfactor Rfree error: 0.004 / Data cutoff high absF: 53281.15 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 60.08 Å2 / ksol: 0.358 e/Å3 | ||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 23.8 Å2
| ||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.15→27.42 Å
| ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.15→2.28 Å / Rfactor Rfree error: 0.012 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS σ(F): 0 / % reflection Rfree: 10 % | ||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 23.8 Å2 | ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.224 / % reflection Rfree: 9.7 % / Rfactor Rwork: 0.175 |
