 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1p4c: High Resolution Structure of Oxidized Active Mutant of (S)-Mandel... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1p4c | ||||||
|---|---|---|---|---|---|---|---|
| Title | High Resolution Structure of Oxidized Active Mutant of (S)-Mandelate Dehydrogenase | ||||||
 Components Components | L(+)-Mandelate Dehydrogenase | ||||||
 Keywords Keywords | OXIDOREDUCTASE / TIM BARREL / Hydroxy acid oxidizing enzyme | ||||||
| Function / homology |  Function and homology information Function and homology information(S)-mandelate dehydrogenase / (S)-mandelate dehydrogenase activity / oxidative photosynthetic carbon pathway / (S)-2-hydroxy-acid oxidase / (S)-2-hydroxy-acid oxidase activity / (R)-mandelate catabolic process / L-lactate dehydrogenase (NAD+) activity / hydrogen peroxide biosynthetic process / aerobic respiration / FMN binding ...(S)-mandelate dehydrogenase / (S)-mandelate dehydrogenase activity / oxidative photosynthetic carbon pathway / (S)-2-hydroxy-acid oxidase / (S)-2-hydroxy-acid oxidase activity / (R)-mandelate catabolic process / L-lactate dehydrogenase (NAD+) activity / hydrogen peroxide biosynthetic process / aerobic respiration / FMN binding / peroxisome / plasma membrane Similarity search - Function | ||||||
| Biological species |  Pseudomonas putida (bacteria) Pseudomonas putida (bacteria) Spinacia oleracea (spinach) Spinacia oleracea (spinach) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.35 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.35 Å | ||||||
 Authors Authors | Sukumar, N. / Mitra, B. / Mathews, F.S. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2004 Title: High Resolution Structures of an Oxidized and Reduced Flavoprotein: THE WATER SWITCH IN A SOLUBLE FORM OF (S)-MANDELATE DEHYDROGENASE Authors: Sukumar, N. / Dewanti, A.R. / Mitra, B. / Mathews, F.S. #1: Journal: Biochemistry / Year: 2001Title: Structure of an active Soluble Mutant of the Membrane- Associated (S)-Mandelate Dehydrogenase Authors: Sukumar, N. / Xu, Y. / Gatti, D.L. / Mitra, B. / Mathews, F.S. | ||||||
| History |
| ||||||
| Remark 999 | SEQUENCE Chimeric mutant of (s)-mandelate dehydrogenase with residues 177-196 replaced by residues ...SEQUENCE Chimeric mutant of (s)-mandelate dehydrogenase with residues 177-196 replaced by residues 176- 195 of glycolate oxidase. The authors believe that SwissProt entry P20932 incorrectly lists residue 15 as ARG instead of ALA. Sequencing of (s)-mandelate dehydrogenase, as well as of the original clone pSCR4, has been done a number of times including site-specific mutants and has repeatedly confirmed that it is ALA 15 rather than ARG 15. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1p4c.cif.gz | 95.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1p4c.ent.gz | 70.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1p4c.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/p4/1p4cftp://data.pdbj.org/pub/pdb/validation_reports/p4/1p4c | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1p5bC  1huvS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 42132.273 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Details: 20 RESIDUE SUBSTITUTION FROM GLYCOLATE OXIDASE AT RESIDUE 177 Source: (gene. exp.) Pseudomonas putida (bacteria), (gene. exp.) Spinacia oleracea (spinach)Production host: |
|---|---|
| #2: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 |
| #3: Chemical | ChemComp-FMN / 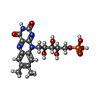  Mass: 456.344 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H21N4O9P Mass: 456.344 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H21N4O9P |
| #4: Chemical | ChemComp-MES /   Mass: 195.237 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H13NO4S / Comment: pH buffer*YM Mass: 195.237 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H13NO4S / Comment: pH buffer*YM |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 400 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 400 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.56 Å3/Da / Density % sol: 52.82 % | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 6.2 Details: ammonium sulfate, ethylene glycol, FMN, Sodium Chloride, pH 6.2, VAPOR DIFFUSION, HANGING DROP, temperature 293K | |||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop / Details: Sukumar, N., (2001) Biochemistry, 40, 9870. | |||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 19-BM / Wavelength: 0.97625 Å / Beamline: 19-BM / Wavelength: 0.97625 Å |
| Detector | Type: CUSTOM-MADE / Detector: CCD |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97625 Å / Relative weight: 1 |
| Reflection | Resolution: 1.35→50 Å / Num. obs: 92753 / % possible obs: 99.8 % / Biso Wilson estimate: 16.6 Å2 / Rmerge(I) obs: 0.059 |
| Reflection shell | Resolution: 1.35→1.4 Å / Rmerge(I) obs: 0.279 / % possible all: 98.1 |
| Reflection | *PLUS Lowest resolution: 500 Å / Num. obs: 91731 / Redundancy: 5.1 % / Rmerge(I) obs: 0.067 |
| Reflection shell | *PLUS % possible obs: 99.1 % / Rmerge(I) obs: 0.282 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1HUV Resolution: 1.35→39.6 Å / Rfactor Rfree error: 0.002 / Data cutoff high absF: 240668.51 / Data cutoff high rms absF: 240668.51 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 57.3837 Å2 / ksol: 0.352965 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 22.9 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.35→39.6 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.35→1.4 Å / Rfactor Rfree error: 0.007 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 50 Å / % reflection Rfree: 10 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
