 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1hkq: PPS10 plasmid DNA replication initiator protein RepA. Replication... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1hkq | ||||||
|---|---|---|---|---|---|---|---|
| Title | PPS10 plasmid DNA replication initiator protein RepA. Replication inactive, dimeric N-terminal domain. | ||||||
 Components Components | REPLICATION PROTEIN | ||||||
 Keywords Keywords | DNA BINDING PROTEIN / WINGED-HELIX / PPS10 PLASMID / REPLICATION INITIATOR DIMER. | ||||||
| Function / homology |  Function and homology information Function and homology informationDNA replication initiation / DNA-directed DNA polymerase activity / identical protein binding Similarity search - Function | ||||||
| Biological species |  PSEUDOMONAS SAVASTANOI (bacteria) PSEUDOMONAS SAVASTANOI (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.75 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.75 Å | ||||||
 Authors Authors | Giraldo, R. / Fernandez-Tornero, C. / Evans, P.R. / Diaz-Orejas, R. / Romero, A. | ||||||
 Citation Citation | Journal: Nat.Struct.Biol. / Year: 2003 Title: A Conformational Switch between Transcriptional Repression and Replication Initiation in Repa Dimerization Domain Authors: Giraldo, R. / Fernandez-Tornero, C. / Evans, P.R. / Diaz-Orejas, R. / Romero, A. #1: Journal: J.Biol.Chem. / Year: 2003 Title: Structural Changes in Repa, a Plasmid Replication Initiator, Upon Binding to Origin DNA Authors: Diaz-Lopez, T. / Lages-Gonzalo, M. / Serrano-Lopez, A. / Alfonso, C. / Rivas, G. / Diaz-Orejas, R. / Giraldo, R. #2: Journal: Fems Microbiol.Rev. / Year: 2003 Title: Common Domains in the Initiators of DNA Replication in Bacteria, Archaea and Eukarya: Combined Structural, Functional and Phylogenetic Perspectives Authors: Giraldo, R. #3: Journal: Embo J. / Year: 1998 Title: Protein Domains and Conformational Changes in the Activation of Repa, a DNA Replication Initiator Authors: Giraldo, R. / Andreu, J.M. / Diaz-Orejas, R. #4: Journal: J.Mol.Biol. / Year: 1992 Title: Genetic and Functional Analysis of the Basic Replicon of Pps10, a Plasmid Specific for Pseudomonas Isolated from Pseudomonas Syringae, Patovar Savastanoi Authors: Nieto, C. / Giraldo, R. / Fernandez-Tresguerres, M.E. / Diaz, R. | ||||||
| History |
| ||||||
| Remark 650 | HELIX DETERMINATION METHOD: AUTHOR PROVIDED. | ||||||
| Remark 700 | SHEET DETERMINATION METHOD: AUTHOR PROVIDED. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1hkq.cif.gz | 65.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1hkq.ent.gz | 48.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1hkq.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hk/1hkqftp://data.pdbj.org/pub/pdb/validation_reports/hk/1hkq | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj













- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (-0.05164, 0.99822, 0.02989), Vector: |
-Components
| #1: Protein | Mass: 15180.649 Da / Num. of mol.: 2 / Fragment: N-TERMINAL DOMAIN DIMER, RESIDUES 2-133 Source method: isolated from a genetically manipulated source Details: PPS10 PLASMID DNA REPLICATION INITIATOR, REPLICATION INACTIVE, DIMERIC SPECIES Source: (gene. exp.) PSEUDOMONAS SAVASTANOI (bacteria) / Plasmid: PRG-RECA-NHIS / Production host: #2: Chemical | ChemComp-HG /   Mass: 200.590 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Hg Mass: 200.590 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Hg#3: Chemical | 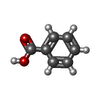  Mass: 122.121 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C7H6O2 Mass: 122.121 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C7H6O2#4: Chemical | ChemComp-PO4 / |   Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4 Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 30 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 30 / Source method: isolated from a natural source / Formula: H2OSequence details | CLONED REPA STARTS WITH A SINGLE MET RESIDUE | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.26 Å3/Da / Density % sol: 45.17 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / pH: 6.2 Details: VAPOUR DIFFUSION (HANGING DROPS), WELLS: 500 MICROLITRES (14% PEG4000, 6% MPD), DROPS: 5 MICROLITRES PROTEIN (5 MG/ML IN 50 MM K2HPO4/ KH2PO4, PH= 6.2),5 MICROLITRES WELL SOL. PLUS 1 ...Details: VAPOUR DIFFUSION (HANGING DROPS), WELLS: 500 MICROLITRES (14% PEG4000, 6% MPD), DROPS: 5 MICROLITRES PROTEIN (5 MG/ML IN 50 MM K2HPO4/ KH2PO4, PH= 6.2),5 MICROLITRES WELL SOL. PLUS 1 MICROLITRE OF P-CL-MERCURIBENZOATE (=1 MM IN WATER), THIN LARGE CRYSTAL PRISMS APPEAR AT 295 DEGREES KELVIN IN 1 MONTH., pH 6.20 | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 295 K / pH: 6.2 / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: X31 / Wavelength: 1.0090, 1.0120 / Beamline: X31 / Wavelength: 1.0090, 1.0120 | |||||||||
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Jul 15, 2000 | |||||||||
| Radiation | Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||
| Radiation wavelength |
| |||||||||
| Reflection | Resolution: 2.75→48.2 Å / Num. obs: 8343 / % possible obs: 97.7 % / Observed criterion σ(I): 2.1 / Redundancy: 5.2 % / Biso Wilson estimate: 45.9 Å2 / Rmerge(I) obs: 0.081 / Net I/σ(I): 8 | |||||||||
| Reflection shell | Resolution: 2.75→2.9 Å / Redundancy: 5.3 % / Rmerge(I) obs: 0.32 / Mean I/σ(I) obs: 2.1 / % possible all: 97.8 | |||||||||
| Reflection | *PLUS Highest resolution: 2.75 Å / Lowest resolution: 48.2 Å / Num. measured all: 44839 / Rmerge(I) obs: 0.081 | |||||||||
| Reflection shell | *PLUS % possible obs: 97.8 % / Rmerge(I) obs: 0.32 / Mean I/σ(I) obs: 2.4 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 2.75→27.89 Å / Rfactor Rfree error: 0.01 / Data cutoff high absF: 1025607.14 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 Details: THE ELECTRON DENSITY FOR RESIDUES SER B36 - LYS B42 IN CHAIN B IS DISCONTINUOUS, INTERPRETED TO BE PARTIALLY DISORDERED. THUS IT HAS BEEN MODELLED BASED ON STEREOCHEMICAL RESTRAINTS.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT / Bsol: 27.2205 Å2 / ksol: 0.305183 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 48.1 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.75→27.89 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.75→2.92 Å / Rfactor Rfree error: 0.034 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Lowest resolution: 2.9 Å |
