 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1hfb: Crystal structure of the tyrosine-regulated 3-deoxy-D-arabino-hep... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1hfb | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the tyrosine-regulated 3-deoxy-D-arabino-heptulosonate-7-phosphate synthase from Saccharomyces cerevisiae complexed with phosphoenolpyruvate | ||||||
 Components Components | TYROSINE-REGULATED 3-DEOXY-D-ARABINO-HEPTULOSONATE-7-PHOSPHATE SYNTHASE | ||||||
 Keywords Keywords | LYASE / BETA-ALPHA-BARREL | ||||||
| Function / homology |  Function and homology information Function and homology information3-deoxy-7-phosphoheptulonate synthase / 3-deoxy-7-phosphoheptulonate synthase activity / chorismate biosynthetic process / aromatic amino acid biosynthetic process / amino acid biosynthetic process / nucleus / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å | ||||||
 Authors Authors | Schneider, T.R. / Hartmann, M. / Braus, G.H. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 2003 Title: Evolution of Feedback-Inhibited Beta /Alpha Barrel Isoenzymes by Gene Duplication and a Single Mutation Authors: Hartmann, M. / Schneider, T.R. / Pfeil, A. / Heinrich, G. / Lipscomb, W. / Braus, G.H. | ||||||
| History |
| ||||||
| Remark 700 | SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS AB, BA, CB, DA, EB, FA, GB, AND HA BELOW ... SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS AB, BA, CB, DA, EB, FA, GB, AND HA BELOW ARE ACTUALLY 8-STRANDED BARRELS. THESE ARE REPRESENTED BY A 9-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1hfb.cif.gz | 530 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1hfb.ent.gz | 436.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1hfb.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hf/1hfbftp://data.pdbj.org/pub/pdb/validation_reports/hf/1hfb | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1qr7S S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given / Matrix: (1),| Details | THE 8 MOLECULES CONTAINED IN THE ASSYMETRIC UNIT AREARRANGED IN TWO TETRAMERS (DIMER OF DIMERS) WITH APPROXIMATE222 POINT SYMMETRY. THE TETRAMERS HAVE VERY SIMILAR ORIENTATIONS,WITH THEIR CENTERS OF MASS BEING RELATED BY ACRYSTALLOGRAPHIC TRANSLATION OF (0.5,0.5,0). | |
-Components
| #1: Protein | Mass: 39797.055 Da / Num. of mol.: 8 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Strain: RH1326 / Production host: #2: Chemical | ChemComp-PEP / 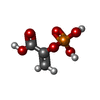  Mass: 168.042 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C3H5O6P Mass: 168.042 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C3H5O6P#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 1251 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 1251 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.22 Å3/Da / Density % sol: 44.69 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 9 / Details: pH 9.00 | ||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 21 ℃ / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: X11 / Wavelength: 0.9076 / Beamline: X11 / Wavelength: 0.9076 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Sep 1, 1998 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9076 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→40 Å / Num. obs: 210365 / % possible obs: 96.3 % / Observed criterion σ(I): -3 / Redundancy: 2.01 % / Rmerge(I) obs: 0.037 |
| Reflection | *PLUS Redundancy: 2.9 % / Num. measured all: 423490 |
| Reflection shell | *PLUS Highest resolution: 1.9 Å / Lowest resolution: 1.97 Å / % possible obs: 89.3 % / Redundancy: 1.2 % / Rmerge(I) obs: 0.354 / Mean I/σ(I) obs: 2.2 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: CHAINS A AND B OF PDB ENTRY 1QR7 Resolution: 1.9→40 Å / Data cutoff high absF: 10000 / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 41.9802 Å2 / ksol: 0.34361 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→40 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor Rfree: 0.261 / Rfactor Rwork: 0.208 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
