 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 1h3o | ||||||
|---|---|---|---|---|---|---|---|
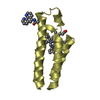
| タイトル | Crystal Structure of the Human TAF4-TAF12 (TAFII135-TAFII20) Complex | ||||||
 要素 要素 |
| ||||||
 キーワード キーワード | TRANSCRIPTION/TBP-ASSOCIATED FACTORS / TBP-ASSOCIATED FACTORS / TFIID / RNA POLYMERASE II TRANSCRIPTION / HISTONE FOLD DOMAINS / NUCLEAR PROTEIN / TRANSCRIPTION-TBP-ASSOCIATED FACTORS complex | ||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報transcription factor TFTC complex / SAGA complex / transcription factor TFIID complex / HIV Transcription Initiation / RNA Polymerase II HIV Promoter Escape / Transcription of the HIV genome / RNA Polymerase II Promoter Escape / RNA Polymerase II Transcription Pre-Initiation And Promoter Opening / RNA Polymerase II Transcription Initiation / RNA Polymerase II Transcription Initiation And Promoter Clearance ...transcription factor TFTC complex / SAGA complex / transcription factor TFIID complex / HIV Transcription Initiation / RNA Polymerase II HIV Promoter Escape / Transcription of the HIV genome / RNA Polymerase II Promoter Escape / RNA Polymerase II Transcription Pre-Initiation And Promoter Opening / RNA Polymerase II Transcription Initiation / RNA Polymerase II Transcription Initiation And Promoter Clearance / regulation of RNA splicing / aryl hydrocarbon receptor binding / MLL1 complex / positive regulation of transcription initiation by RNA polymerase II / regulation of DNA repair / RNA polymerase II preinitiation complex assembly / ovarian follicle development / RNA Polymerase II Pre-transcription Events / TBP-class protein binding / male germ cell nucleus / transcription initiation at RNA polymerase II promoter / DNA-templated transcription initiation / mRNA transcription by RNA polymerase II / HATs acetylate histones / transcription by RNA polymerase II / DNA-binding transcription factor binding / Regulation of TP53 Activity through Phosphorylation / transcription coactivator activity / protein heterodimerization activity / regulation of transcription by RNA polymerase II / regulation of DNA-templated transcription / positive regulation of DNA-templated transcription / chromatin / positive regulation of transcription by RNA polymerase II / protein-containing complex / DNA binding / nucleoplasm / nucleus / cytosol 類似検索 - 分子機能 | ||||||
| 生物種 |  HOMO SAPIENS (ヒト) HOMO SAPIENS (ヒト) | ||||||
| 手法 |  X線回折 / シンクロトロン / 多波長異常分散 / 解像度: 2.3 Å X線回折 / シンクロトロン / 多波長異常分散 / 解像度: 2.3 Å | ||||||
 データ登録者 データ登録者 | Werten, S. / Mitschler, A. / Moras, D. | ||||||
 引用 引用 | ジャーナル: J.Biol.Chem. / 年: 2002 タイトル: Crystal Structure of a Subcomplex of Human Transcription Factor TFIID Formed by TATA Binding Protein-Associated Factors Htaf4 (Htaf(II)135) and Htaf12 (Htaf(II)20). 著者: Werten, S. / Mitschler, A. / Romier, C. / Gangloff, Y.-G. / Thuault, S. / Davidson, I. / Moras, D. #1: ジャーナル: Mol.Cell.Biol. / 年: 2000 タイトル: The Human TFIID Components Tafii135 and Tafii20 and the Yeast Saga Components Ada1 and Tafii68 Heterodimerize to Form Histone-Like Pairs 著者: Gangloff, Y.-G. / Werten, S. / Romier, C. / Carre, L. / Poch, O. / Moras, D. / Davidson, I. | ||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 1h3o.cif.gz | 67 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb1h3o.ent.gz | 50.7 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 1h3o.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/h3/1h3oftp://data.pdbj.org/pub/pdb/validation_reports/h3/1h3o | HTTPS FTP |
|---|
-関連構造データ









-リンク
 PDBj
PDBj

- 集合体
集合体
| 登録構造単位 | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 単位格子 |
| ||||||||
| 非結晶学的対称性 (NCS) | NCS oper: (Code: given Matrix: (0.96526, -0.00695, 0.26119), ベクター: 詳細 | TWO HETERODIMERS FORMED BY CHAINS A AND B , OR CHAINSC AND D. | |
-要素
| #1: タンパク質 | 分子量: 8813.803 Da / 分子数: 2 / 断片: HISTONE FOLD DOMAIN, RESIDUES 870-943 / 変異: YES / 由来タイプ: 組換発現 詳細: N-TERMINAL SELENOMETHIONINE INSERT, UNIFORM SELENO-METHIONINE LABELING 由来: (組換発現) HOMO SAPIENS (ヒト) / 解説: SYNTHETIC GENE / プラスミド: PACYC-11B / 発現宿主:  #2: タンパク質 | 分子量: 9053.836 Da / 分子数: 2 / 断片: HISTONE FOLD DOMAIN, RESIDUES 57-128 / 変異: YES / 由来タイプ: 組換発現 詳細: RESIDUES (GLY SER HIS MSE) INSERTED AT THE N-TERMINUS, REMAINDER OF HISTIDINE-TAG AFTER THROMBIN TREATMENT, UNIFORM SELENO-METHIONINE LABELING 由来: (組換発現) HOMO SAPIENS (ヒト) / 解説: SYNTHETIC GENE / プラスミド: PET-15B / 発現宿主: #3: 水 | ChemComp-HOH / |  分子量: 18.015 Da / 分子数: 158 / 由来タイプ: 天然 / 式: H2O 分子量: 18.015 Da / 分子数: 158 / 由来タイプ: 天然 / 式: H2O構成要素の詳細 | HTAF4: MULTIMERIC PROTEIN COMPLEX THAT PLAYS A CENTRAL ROLE IN MEDIATING PROMOTER RESPONSES TO ...HTAF4: MULTIMERIC | Has protein modification | Y | 配列の詳細 | ENGINEERED DELETION MUTANT THE HTAF12 POLYPEPTIDE THAT WAS USED FOR CRYSTALLIZATION CONTAINED THE ...ENGINEERED | |
|---|
-実験情報
-実験
| 実験 | 手法: X線回折 / 使用した結晶の数: 1 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 2.2 Å3/Da / 溶媒含有率: 42 % 解説: DATA QUALITY STATISTICS LISTED PERTAIN TO REMOTE WAVELENGTH DATA (0.90730 A) | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 結晶化 | pH: 5.25 / 詳細: pH 5.25 | ||||||||||||||||||||||||||||||
| 結晶化 | *PLUS 温度: 24 ℃ / 手法: 蒸気拡散法, ハンギングドロップ法 | ||||||||||||||||||||||||||||||
| 溶液の組成 | *PLUS
|
-データ収集
| 回折 | 平均測定温度: 100 K | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 放射光源 | 由来: シンクロトロン / サイト: EMBL/DESY, HAMBURG  / ビームライン: BW7A / 波長: 0.90730,0.97740 / ビームライン: BW7A / 波長: 0.90730,0.97740 | |||||||||
| 検出器 | タイプ: MARRESEARCH / 検出器: CCD / 日付: 2001年6月9日 / 詳細: PREMIRROR, BENT MIRROR | |||||||||
| 放射 | モノクロメーター: DOUBLE CRYSTAL FOCUSSING MONOCHROMATOR プロトコル: MAD / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray | |||||||||
| 放射波長 |
| |||||||||
| 反射 | 解像度: 2.3→20 Å / Num. obs: 24237 / % possible obs: 92.3 % / 冗長度: 1.5 % / Biso Wilson estimate: 21.9 Å2 / Rsym value: 0.034 / Net I/σ(I): 19.9 | |||||||||
| 反射 シェル | 解像度: 2.3→2.38 Å / 冗長度: 1.5 % / Mean I/σ(I) obs: 4.45 / Rsym value: 0.167 / % possible all: 90.1 | |||||||||
| 反射 | *PLUS 最高解像度: 2.3 Å / Num. obs: 25898 / % possible obs: 98.9 % / Num. measured all: 61754 / Rmerge(I) obs: 0.035 | |||||||||
| 反射 シェル | *PLUS % possible obs: 99.7 % / Num. unique obs: 2588 / Num. measured obs: 6073 / Rmerge(I) obs: 0.122 / Mean I/σ(I) obs: 7.56 |
- 解析
解析
| ソフトウェア |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 構造決定の手法: 多波長異常分散 / 解像度: 2.3→19.63 Å / Rfactor Rfree error: 0.011 / Data cutoff high absF: 1965882.6 / Isotropic thermal model: RESTRAINED / 交差検証法: THROUGHOUT / σ(F): 0 / 立体化学のターゲット値: MLF 詳細: NO ELECTRON DENSITY WAS OBSERVED FOR THE C-TERMINAL PORTION OF HTAF4, RESIDUES 918-943, WHICH IS THEREFORE ABSENT FROM THE MODEL. THE FACT THAT PART OF THE PROTEIN COMPLEX DID NOT SHOW UP IN ...詳細: NO ELECTRON DENSITY WAS OBSERVED FOR THE C-TERMINAL PORTION OF HTAF4, RESIDUES 918-943, WHICH IS THEREFORE ABSENT FROM THE MODEL. THE FACT THAT PART OF THE PROTEIN COMPLEX DID NOT SHOW UP IN ELECTRON DENSITY MAPS WAS NOT DUE TO PROTEOLYSIS (AS EVIDENCED BY MASS SPECTROSCOPY OF REDISSOLVED CRYSTALS), INDICATING THAT THE REGIONS INVOLVED ARE DISORDERED WITHIN THE CRYSTAL LATTICE.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶媒の処理 | 溶媒モデル: FLAT MODEL / Bsol: 58.9338 Å2 / ksol: 0.357292 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | Biso mean: 47.4 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化ステップ | サイクル: LAST / 解像度: 2.3→19.63 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS精密化 シェル | 解像度: 2.3→2.44 Å / Rfactor Rfree error: 0.035 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化 | *PLUS 最低解像度: 20 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶媒の処理 | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 | *PLUS
|
