 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1h12 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of a cold-adapted family 8 xylanase | |||||||||
 Components Components | ENDO-1,4-BETA-XYLANASE | |||||||||
 Keywords Keywords | HYDROLASE / XYLAN DEGRADATION / PSYCHROPHILIC / COLD ADAPTATION / TEMPERATURE / GLYCOSYL HYDROLASE / FAMILY 8 | |||||||||
| Function / homology |  Function and homology information Function and homology informationcellulase / cellulase activity / xylan catabolic process / cellulose catabolic process Similarity search - Function | |||||||||
| Biological species |  PSEUDOALTEROMONAS HALOPLANKTIS (bacteria) PSEUDOALTEROMONAS HALOPLANKTIS (bacteria) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.2 Å | |||||||||
 Authors Authors | Van Petegem, F. / Collins, T. / Meuwis, M.A. / Feller, G. / Gerday, C. / Van Beeumen, J. | |||||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2003 Title: The Structure of a Cold-Adapted Family 8 Xylanase at 1.3 A Resolution: Structural Adaptations to Cold and Investigation of the Active Site Authors: Van Petegem, F. / Collins, T. / Meuwis, M.A. / Gerday, C. / Feller, G. / Van Beeumen, J. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1h12.cif.gz | 185.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1h12.ent.gz | 146.6 KB | Display | PDB format |
| PDBx/mmJSON format | 1h12.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/h1/1h12ftp://data.pdbj.org/pub/pdb/validation_reports/h1/1h12 | HTTPS FTP |
|---|
-Related structure data












-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 46024.770 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) PSEUDOALTEROMONAS HALOPLANKTIS (bacteria)Production host: |
|---|---|
| #2: Sugar | ChemComp-XYP / 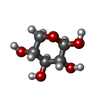  Type: D-saccharide, beta linking / Mass: 150.130 Da / Num. of mol.: 1 Type: D-saccharide, beta linking / Mass: 150.130 Da / Num. of mol.: 1Source method: isolated from a genetically manipulated source Formula: C5H10O5 |
| #3: Sugar | ChemComp-XYS / 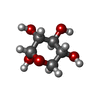  Type: D-saccharide, alpha linking / Mass: 150.130 Da / Num. of mol.: 1 Type: D-saccharide, alpha linking / Mass: 150.130 Da / Num. of mol.: 1Source method: isolated from a genetically manipulated source Formula: C5H10O5 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 390 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 390 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.43 Å3/Da / Density % sol: 47.5 % Description: CRYO-ANNEALING LOWERS THE MOSAICITY AND INCREASES THE RESOLUTION LIMIT | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 7 Details: TAKE 2 - 10 MG/ML PROTEIN IN 20MM MOPS, 50MM NACL, 2% TREHALOSE, PH 7.5. ADD EQUAL VOLUME OF 70% MPD, 0.1M PHOSPHATE BUFFER PH 7.0 WITH 10MG/ML XYLOBIOSE IN A HANGING DROP EXPERIMENT AT 4 DEGREES CENTIGRADE. | ||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 5 / Method: vapor diffusion, hanging dropDetails: Van Petegem, F., (2002) Acta Crystallogr., D58, 1494. | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ELETTRA  / Beamline: 5.2R / Wavelength: 1 / Beamline: 5.2R / Wavelength: 1 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.2→20 Å / Num. obs: 141707 / % possible obs: 99.4 % / Observed criterion σ(I): 2 / Redundancy: 5 % / Rmerge(I) obs: 0.032 / Net I/σ(I): 43.52 |
| Reflection shell | Resolution: 1.2→1.22 Å / Redundancy: 2.5 % / Rmerge(I) obs: 0.235 / Mean I/σ(I) obs: 3.69 / % possible all: 91.9 |
| Reflection | *PLUS Highest resolution: 1.2 Å / Lowest resolution: 20 Å / Num. measured all: 1757738 |
| Reflection shell | *PLUS Highest resolution: 1.2 Å / % possible obs: 91.9 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: WILD TYPE WITHOUT LIGAND Resolution: 1.2→65.94 Å / Cor.coef. Fo:Fc: 0.985 / Cor.coef. Fo:Fc free: 0.977 / SU B: 0.752 / SU ML: 0.018 / Cross valid method: THROUGHOUT / ESU R: 0.025 / ESU R Free: 0.026 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 10.66 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.2→65.94 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|