 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1gxf: CRYSTAL STRUCTURE OF TRYPANOSOMA CRUZI TRYPANOTHIONE REDUCTASE IN... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1gxf | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF TRYPANOSOMA CRUZI TRYPANOTHIONE REDUCTASE IN COMPLEX WITH THE INHIBITOR QUINACRINE MUSTARD | ||||||
 Components Components | TRYPANOTHIONE REDUCTASE (OXIDIZED FORM) | ||||||
 Keywords Keywords | OXIDOREDUCTASE / TRYPANOTHIONE REDUCTASE / FAD DEPENDENT / DISULPHIDE OXIDOREDUCTASE / QUINACRINE MUSTARD / INHIBITOR / REDOX-ACTIVE CENTER / FLAVOPROTEIN / FAD / NADP | ||||||
| Function / homology |  Function and homology information Function and homology informationtrypanothione-disulfide reductase / trypanothione-disulfide reductase (NADPH) activity / glutathione-disulfide reductase (NADPH) activity / cell redox homeostasis / glutathione metabolic process / flavin adenine dinucleotide binding / cellular response to oxidative stress / mitochondrion / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.7 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.7 Å | ||||||
 Authors Authors | Bond, C.S. / Peterson, M.R. / Vickers, T.J. / Fairlamb, A.H. / Hunter, W.N. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2004 Title: Two Interacting Binding Sites for Quinacrine Derivatives in the Active Site of Trypanothione Reductase: A Template for Drug Design Authors: Saravanamuthu, A. / Vickers, T.J. / Bond, C.S. / Peterson, M.R. / Hunter, W.N. / Fairlamb, A.H. | ||||||
| History |
| ||||||
| Remark 700 | SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN ... SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN THE SHEET RECORDS BELOW, TWO SHEETS ARE DEFINED. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1gxf.cif.gz | 203.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1gxf.ent.gz | 162.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1gxf.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gx/1gxfftp://data.pdbj.org/pub/pdb/validation_reports/gx/1gxf | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1tytS S: Starting model for refinement |
|---|---|
| Similar structure data |







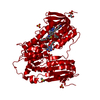







-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (-0.979689, 0.200521, 0.000849), Vector: |
-Components
| #1: Protein | Mass: 53925.668 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Details: COVALENTLY LABELLED WITH QUINACRINE MUSTARD / Source: (gene. exp.)  #2: Chemical |   Mass: 785.550 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C27H33N9O15P2 / Comment: FAD*YM Mass: 785.550 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C27H33N9O15P2 / Comment: FAD*YM#3: Chemical | ChemComp-MAE / | 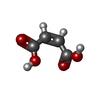  Mass: 116.072 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H4O4 Mass: 116.072 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H4O4#4: Chemical | ChemComp-QUM /   Mass: 468.847 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C23H28Cl3N3O Mass: 468.847 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C23H28Cl3N3O#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 53 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 53 / Source method: isolated from a natural source / Formula: H2OCompound details | TRYPANOTHIONE IS THE PARASITE ANALOG OF GLUTATHIONE AND THIS ENZYME IS THE EQUIVALENT OF ...TRYPANOTHI | Has protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.17 Å3/Da / Density % sol: 61 % |
|---|---|
| Crystal grow | pH: 6 / Details: MALEIC BUFFER, PEG 4000, pH 6.00 |
-Data collection
| Diffraction | Mean temperature: 293 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: BM14 / Wavelength: 0.92 / Beamline: BM14 / Wavelength: 0.92 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.92 Å / Relative weight: 1 |
| Reflection | Resolution: 2.7→21 Å / Num. obs: 29073 / % possible obs: 79 % / Observed criterion σ(I): 0 / Redundancy: 3 % / Rmerge(I) obs: 0.08 / Net I/σ(I): 9.4 |
| Reflection shell | Resolution: 2.7→2.9 Å / Rmerge(I) obs: 0.34 / Mean I/σ(I) obs: 2.6 / % possible all: 82 |
- Processing
Processing
| Software |
| ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1TYT Resolution: 2.7→21 Å / SU B: 11.848 / SU ML: 0.25126 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R Free: 0.3924 Details: THE MISSING N- AND C-TERMINAL RESIDUES COULD NOT BE SEEN IN THE ELECTRON DENSITY MAP CLEARLY
| ||||||||||||||||
| Displacement parameters | Biso mean: 46 Å2 | ||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.7→21 Å
|
