 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1gja: ENGINEERING INHIBITORS HIGHLY SELECTIVE FOR THE S1 SITES OF SER19... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1gja | ||||||
|---|---|---|---|---|---|---|---|
| Title | ENGINEERING INHIBITORS HIGHLY SELECTIVE FOR THE S1 SITES OF SER190 TRYPSIN-LIKE SERINE PROTEASE DRUG TARGETS | ||||||
 Components Components | (UROKINASE-TYPE PLASMINOGEN ACTIVATOR) x 2 | ||||||
 Keywords Keywords | BLOOD CLOTTING / hydrolase / selectivity at S1 / H2O displacement / uPA / tPA / Ser190/Ala190 protease / structure-based drug design | ||||||
| Function / homology |  Function and homology information Function and homology informationu-plasminogen activator / regulation of smooth muscle cell-matrix adhesion / urokinase plasminogen activator signaling pathway / regulation of plasminogen activation / regulation of fibrinolysis / regulation of wound healing / protein complex involved in cell-matrix adhesion / regulation of signaling receptor activity / negative regulation of plasminogen activation / serine-type endopeptidase complex ...u-plasminogen activator / regulation of smooth muscle cell-matrix adhesion / urokinase plasminogen activator signaling pathway / regulation of plasminogen activation / regulation of fibrinolysis / regulation of wound healing / protein complex involved in cell-matrix adhesion / regulation of signaling receptor activity / negative regulation of plasminogen activation / serine-type endopeptidase complex / regulation of smooth muscle cell migration / Dissolution of Fibrin Clot / smooth muscle cell migration / plasminogen activation / regulation of cell adhesion mediated by integrin / tertiary granule membrane / negative regulation of fibrinolysis / regulation of cell adhesion / specific granule membrane / serine protease inhibitor complex / fibrinolysis / chemotaxis / blood coagulation / regulation of cell population proliferation / response to hypoxia / positive regulation of cell migration / external side of plasma membrane / serine-type endopeptidase activity / focal adhesion / Neutrophil degranulation / cell surface / signal transduction / proteolysis / extracellular space / extracellular exosome / extracellular region / plasma membrane Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / FOURIER SYNTHESIS / Resolution: 1.56 Å X-RAY DIFFRACTION / FOURIER SYNTHESIS / Resolution: 1.56 Å | ||||||
 Authors Authors | Katz, B.A. / Sprengeler, P.A. / Luong, C. / Verner, E. / Spencer, J.R. / Breitenbucher, J.G. / Hui, H. / McGee, D. / Allen, D. / Martelli, A. / Mackman, R.L. | ||||||
 Citation Citation | Journal: Chem.Biol. / Year: 2001 Title: Engineering inhibitors highly selective for the S1 sites of Ser190 trypsin-like serine protease drug targets. Authors: Katz, B.A. / Sprengeler, P.A. / Luong, C. / Verner, E. / Elrod, K. / Kirtley, M. / Janc, J. / Spencer, J.R. / Breitenbucher, J.G. / Hui, H. / McGee, D. / Allen, D. / Martelli, A. / Mackman, R.L. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1gja.cif.gz | 129.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1gja.ent.gz | 102.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1gja.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 1gja_validation.pdf.gz | 707.1 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 1gja_full_validation.pdf.gz | 717.4 KB | Display | |
| Data in XML | 1gja_validation.xml.gz | 16.9 KB | Display | |
| Data in CIF | 1gja_validation.cif.gz | 24.2 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gj/1gjaftp://data.pdbj.org/pub/pdb/validation_reports/gj/1gja | HTTPS FTP |
-Related structure data
| Related structure data |  1gj4C  1gj5C  1gj6C  1gj7C  1gj8C  1gj9C  1gjbC  1gjcC  1gjdC C: citing same article ( |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||
| Unit cell |
| ||||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein/peptide | Mass: 2708.183 Da / Num. of mol.: 1 / Fragment: SHORT CHAIN Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Description: P.PASTORIS / Plasmid: PPIC9LMWUPA / Production host:  | ||||||
|---|---|---|---|---|---|---|---|
| #2: Protein | Mass: 28435.428 Da / Num. of mol.: 1 / Fragment: CATALYTIC DOMAIN / Mutation: N145A Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Description: P.PASTORIS / Plasmid: PPIC9LMWUPA / Production host: | ||||||
| #3: Chemical |   Mass: 192.124 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H8O7 Mass: 192.124 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H8O7#4: Chemical | ChemComp-135 / | 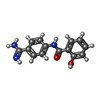  Mass: 255.272 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H13N3O2 Mass: 255.272 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H13N3O2#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 249 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 249 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.01 Å3/Da / Density % sol: 38.94 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion / pH: 6.5 Details: 2-propanol PEG 4000, pH 6.5, vapor diffusion at 298 K, pH 6.50 | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 7.4 / Method: vapor diffusion, hanging drop / Details: Katz, B.A., (2000) Chem.Biol., 7, 299. | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS IV / Detector: IMAGE PLATE / Date: Aug 24, 1998 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.41→41.8 Å / Num. all: 48181 / Num. obs: 27315 / Observed criterion σ(I): 1 / Redundancy: 2.2 % / Rmerge(I) obs: 0.054 / Net I/σ(I): 13.4 |
| Reflection shell | Resolution: 1.6→1.75 Å / Rmerge(I) obs: 0.26 / Num. unique all: 3808 / % possible all: 49.2 |
| Reflection | *PLUS Num. obs: 25786 |
| Reflection shell | *PLUS Highest resolution: 1.56 Å / Lowest resolution: 1.63 Å |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS / Resolution: 1.56→7 Å / Cross valid method: THROUGHOUT / σ(F): 2 / Stereochemistry target values: X-PLOR force field Details: Only Pro_A5 to Thr_A17 are included for the A-chain. Residues prior and after these residues are not visible (disordered). Residues after Lys_B243 are not visible (disordered). Residues ...Details: Only Pro_A5 to Thr_A17 are included for the A-chain. Residues prior and after these residues are not visible (disordered). Residues after Lys_B243 are not visible (disordered). Residues simultaneously refined in two or more conformations are: Met_B47, Met_B81, Ser_B95, Glu_B110B, Leu_B235 Disordered waters are: HOH7 which is close to HOH8 HOH432 which is close to HOH433 HOH588 which is close to HOH589 HOH704 which is close to HOH705 HOH838 which is close to HOH839 HOH780 which is close to HOH781 HOH1081 which is close to HOH1082 HOH1135 which is close to a symmetry-related equivalent of itself; HOH1119 which is close to HOH1120 No energy terms between citrate 1 and 2 are included because they are hydrogen-bonded to one another via an unusually short hydrogen bond between carboxylate / hydroxyl groups.
| ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.56→7 Å
| ||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2 Å / % reflection Rfree: 10 % / Rfactor all: 0.187 / Rfactor obs: 0.159 / Rfactor Rwork: 0.159 | ||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||
| LS refinement shell | *PLUS Highest resolution: 1.56 Å / Lowest resolution: 1.63 Å / Rfactor Rwork: 0.187 / Rfactor obs: 0.187 |
