 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1gbd: ALPHA-LYTIC PROTEASE WITH MET 190 REPLACED BY ALA AND GLY 216 REP... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1gbd | ||||||
|---|---|---|---|---|---|---|---|
| Title | ALPHA-LYTIC PROTEASE WITH MET 190 REPLACED BY ALA AND GLY 216 REPLACED BY ALA COMPLEX WITH METHOXYSUCCINYL-ALA-ALA-PRO-PHENYLALANINE BORONIC ACID | ||||||
 Components Components |
| ||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / ACTIVE-SITE MUTATION / SERINE PROTEINASE / HYDROLASE-HYDROLASE INHIBITOR COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationalpha-lytic endopeptidase / serine-type endopeptidase activity / proteolysis / extracellular region Similarity search - Function | ||||||
| Biological species |  Lysobacter enzymogenes (bacteria) Lysobacter enzymogenes (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.2 Å X-RAY DIFFRACTION / Resolution: 2.2 Å | ||||||
 Authors Authors | Mace, J.E. / Agard, D.A. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 1995 Title: Kinetic and structural characterization of mutations of glycine 216 in alpha-lytic protease: a new target for engineering substrate specificity. Authors: Mace, J.E. / Agard, D.A. #1: Journal: Biochemistry / Year: 1991Title: Structural Basis for Broad Specificity in Alpha-Lytic Protease Authors: Bone, R. / Fujushige, A. / Kettner, C.A. / Agard, D.A. #2: Journal: Nature / Year: 1989Title: Structural Plasticity Broadens the Specificity of an Engineered Protease Authors: Bone, R. / Silen, J.L. / Agard, D.A. #3: Journal: Biochemistry / Year: 1989Title: Structural Analysis of Specificity: Alpha-Lytic Protease Complexes with Analogues of Reaction Intermediates Authors: Bone, R. / Frank, D. / Kettner, D. / Agard, D.A. #4: Journal: Biochemistry / Year: 1987Title: Serine Protease Mechanism: Structure of an Inhibitory Complex of Alpha-Lytic Protease and a Tightly Bound Peptide Boronic Acid Authors: Bone, R. / Shenvi, A.B. / Kettner, C.A. / Agard, D.A. #5: Journal: J.Mol.Biol. / Year: 1985Title: Refined Structure of Alpha-Lytic Protease at 1.7 Angstroms Resolution. Analysis of Hydrogen Bonding and Solvent Structure Authors: Fujinaga, M. / Delbaere, L.T.J. / Brayer, G.D. / James, M.N.G. #6: Journal: J.Mol.Biol. / Year: 1979Title: Molecular Structure of the Alpha-Lytic Protease from Myxobacter 495 at 2.8 Angstroms Resolution Authors: Brayer, G.D. / Delbaere, L.T.J. / James, M.N.G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1gbd.cif.gz | 54.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1gbd.ent.gz | 37.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1gbd.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 1gbd_validation.pdf.gz | 426.6 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 1gbd_full_validation.pdf.gz | 427.5 KB | Display | |
| Data in XML | 1gbd_validation.xml.gz | 11.6 KB | Display | |
| Data in CIF | 1gbd_validation.cif.gz | 16.2 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gb/1gbdftp://data.pdbj.org/pub/pdb/validation_reports/gb/1gbd | HTTPS FTP |
-Related structure data
| Related structure data |  1gbaC  1gbbC  1gbcC  1gbeC  1gbfC  1gbhC  1gbiC  1gbjC  1gbkC  1gblC  1gbmC C: citing same article ( |
|---|---|
| Similar structure data |






-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 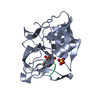
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 19829.041 Da / Num. of mol.: 1 / Mutation: M190A, G216A Source method: isolated from a genetically manipulated source Source: (gene. exp.) Lysobacter enzymogenes (bacteria) / Strain: 495 / Gene: ALPHA-LYTIC PROTEASE PREPROENZ / Plasmid: PALP12 (PBR322-DERIVATIVE) / Gene (production host): ALPHA-LYTIC PROTEASE PREPROENZYME / Production host: | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| #2: Protein/peptide | 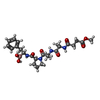  Type: Peptide-like / Class: Inhibitor / Mass: 518.367 Da / Num. of mol.: 1 / Source method: obtained synthetically Type: Peptide-like / Class: Inhibitor / Mass: 518.367 Da / Num. of mol.: 1 / Source method: obtained syntheticallyReferences: O-methylsuccinyl-alanyl-alanyl-prolyl-borophenylalanine | ||||||||
| #3: Chemical |   Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 156 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 156 / Source method: isolated from a natural source / Formula: H2OCompound details | INHIBITORY PEPTIDE BORONIC ACIDS ARE PEPTIDE ANALOGS IN WHICH THE C-TERMINAL CARBOXYL GROUP HAS ...INHIBITORY | Has protein modification | Y | Sequence details | CHAIN A RESIDUE NUMBERING IS DONE BY HOMOLOGY WITH CHYMOTRYPSIN FOR RESIDUES 15A - 245. CHAIN P ...CHAIN A RESIDUE NUMBERING IS DONE BY HOMOLOGY WITH CHYMOTRYPS | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.49 Å3/Da / Density % sol: 50.53 % | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal | *PLUS Density % sol: 48 % | ||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / pH: 8 / Method: vapor diffusion, hanging drop | ||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction source | Wavelength: 1.5418 |
|---|---|
| Detector | Type: RIGAKU AFC-6R / Detector: DIFFRACTOMETER / Date: Feb 16, 1994 |
| Radiation | Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.2→17.5 Å / Num. obs: 10431 / % possible obs: 93 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.2→17.5 Å / σ(F): 2 Details: THE METHOXYSUCCINYL PORTION OF THE INHIBITOR WAS DISORDERED AND NO COORDINATES ARE INCLUDED FOR IT IN THIS ENTRY
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.2→17.5 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
