 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1g0e: SITE-SPECIFIC MUTANT (HIS64 REPLACED WITH ALA) OF HUMAN CARBONIC ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1g0e | ||||||
|---|---|---|---|---|---|---|---|
| Title | SITE-SPECIFIC MUTANT (HIS64 REPLACED WITH ALA) OF HUMAN CARBONIC ANHYDRASE II COMPLEXED WITH 4-METHYLIMIDAZOLE | ||||||
 Components Components | CARBONIC ANHYDRASE II | ||||||
 Keywords Keywords | LYASE / TWISTED BETA SHEET / CHEMICAL RESCUE / 4-METHYLIMIDAZOLE / ZINC METALLOENZYME / PROTEIN LIGAND COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationpositive regulation of dipeptide transmembrane transport / : / regulation of monoatomic anion transport / secretion / cyanamide hydratase / cyanamide hydratase activity / arylesterase activity / regulation of chloride transport / Reversible hydration of carbon dioxide / morphogenesis of an epithelium ...positive regulation of dipeptide transmembrane transport / : / regulation of monoatomic anion transport / secretion / cyanamide hydratase / cyanamide hydratase activity / arylesterase activity / regulation of chloride transport / Reversible hydration of carbon dioxide / morphogenesis of an epithelium / angiotensin-activated signaling pathway / Developmental Lineage of Pancreatic Ductal Cells / carbonic anhydrase / regulation of intracellular pH / carbonate dehydratase activity / positive regulation of synaptic transmission, GABAergic / carbon dioxide transport / Erythrocytes take up oxygen and release carbon dioxide / Erythrocytes take up carbon dioxide and release oxygen / neuron cellular homeostasis / apical part of cell / myelin sheath / extracellular exosome / zinc ion binding / plasma membrane / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 1.6 Å X-RAY DIFFRACTION / Resolution: 1.6 Å | ||||||
 Authors Authors | Duda, D. / McKenna, R. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2001 Title: Structural and kinetic analysis of the chemical rescue of the proton transfer function of carbonic anhydrase II. Authors: Duda, D. / Tu, C. / Qian, M. / Laipis, P. / Agbandje-McKenna, M. / Silverman, D.N. / McKenna, R. #1: Journal: J.Mol.Biol. / Year: 1992Title: Structure of Native and Apo Carbonic Anhydrase II and Structure of Some of its Anion-ligand Complexes. Authors: Hakansson, K. / Carlsson, M. / Svensson, L.A. / Liljas, A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1g0e.cif.gz | 72.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1g0e.ent.gz | 52.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1g0e.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/g0/1g0eftp://data.pdbj.org/pub/pdb/validation_reports/g0/1g0e | HTTPS FTP |
|---|
-Related structure data





















-Links
 PDBj
PDBj














- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | THE BIOLOGICAL ASSEMBLY IS A MONOMER CONSTRUCTED FROM CHAIN A |
-Components
| #1: Protein | Mass: 29221.992 Da / Num. of mol.: 1 / Mutation: H64A Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Tissue: BLOOD / Plasmid: PET / Production host:  | ||
|---|---|---|---|
| #2: Chemical | ChemComp-ZN /   Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn | ||
| #3: Chemical | ChemComp-HG /   Mass: 200.590 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Hg Mass: 200.590 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Hg | ||
| #4: Chemical | ChemComp-4MZ / 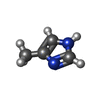  Mass: 82.104 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6N2 Mass: 82.104 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6N2 | ||
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 318 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 318 / Source method: isolated from a natural source / Formula: H2O | ||
| Nonpolymer details | THE LIGAND 4MZ IS CALLED 4MI IN THE PUBLICATIO| Sequence details | RESIDUES 125 AND 127 ARE COVALENTLY | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.09 Å3/Da / Density % sol: 41.09 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 7.8 Details: Ammonium Sulfate, Mercury Chloride, Tris HCl, pH 7.8, VAPOR DIFFUSION, HANGING DROP, temperature 277K | ||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU / Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS / Detector: IMAGE PLATE / Date: Mar 28, 2000 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.6→25 Å / Num. all: 468251 / Num. obs: 28456 / % possible obs: 88.8 % / Observed criterion σ(F): 1 / Observed criterion σ(I): 1 / Redundancy: 16.46 % / Biso Wilson estimate: 9.9 Å2 / Rmerge(I) obs: 0.047 / Net I/σ(I): 15.2 |
| Reflection shell | Resolution: 1.6→25 Å / Redundancy: 16.46 % / Rmerge(I) obs: 0.047 / Num. unique all: 28456 / % possible all: 88.8 |
| Reflection | *PLUS Num. measured all: 468251 |
| Reflection shell | *PLUS Lowest resolution: 1.66 Å / % possible obs: 64.1 % / Num. unique obs: 2018 / Rmerge(I) obs: 0.107 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.6→25 Å / Rfactor Rfree error: 0.005 / Data cutoff high absF: 918664.21 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 1 / σ(I): 1 / Stereochemistry target values: Brunger & Adams
| |||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 47.68 Å2 / ksol: 0.389 e/Å3 | |||||||||||||||||||||||||
| Displacement parameters | Biso mean: 11.2 Å2
| |||||||||||||||||||||||||
| Refine analyze |
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.6→25 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.6→1.7 Å / Rfactor Rfree error: 0.016 / Total num. of bins used: 6
| |||||||||||||||||||||||||
| Xplor file |
| |||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 1 / Classification: refinement | |||||||||||||||||||||||||
| Refinement | *PLUS σ(F): 1 / % reflection Rfree: 4.9 % / Rfactor all: 0.18 / Rfactor obs: 179 / Rfactor Rfree: 0.2 | |||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 11.2 Å2 | |||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| |||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.207 / % reflection Rfree: 4.4 % / Rfactor Rwork: 0.188 |
