 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1fk5: STRUCTURAL BASIS OF NON-SPECIFIC LIPID BINDING IN MAIZE LIPID-TRA... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1fk5 | ||||||
|---|---|---|---|---|---|---|---|
| Title | STRUCTURAL BASIS OF NON-SPECIFIC LIPID BINDING IN MAIZE LIPID-TRANSFER PROTEIN COMPLEXES WITH OLEIC ACID REVEALED BY HIGH-RESOLUTION X-RAY CRYSTALLOGRAPHY | ||||||
 Components Components | NONSPECIFIC LIPID-TRANSFER PROTEIN | ||||||
 Keywords Keywords | LIPID TRANSPORT / protein-lipid complex | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / molecular replacement / Resolution: 1.3 Å X-RAY DIFFRACTION / SYNCHROTRON / molecular replacement / Resolution: 1.3 Å | ||||||
 Authors Authors | Han, G.W. / Lee, J.Y. / Song, H.K. / Shin, D.H. / Suh, S.W. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2001 Title: Structural basis of non-specific lipid binding in maize lipid-transfer protein complexes revealed by high-resolution X-ray crystallography. Authors: Han, G.W. / Lee, J.Y. / Song, H.K. / Chang, C. / Min, K. / Moon, J. / Shin, D.H. / Kopka, M.L. / Sawaya, M.R. / Yuan, H.S. / Kim, T.D. / Choe, J. / Lim, D. / Moon, H.J. / Suh, S.W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1fk5.cif.gz | 30.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1fk5.ent.gz | 20.6 KB | Display | PDB format |
| PDBx/mmJSON format | 1fk5.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/fk/1fk5ftp://data.pdbj.org/pub/pdb/validation_reports/fk/1fk5 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1fk0C  1fk1C  1fk2C  1fk3C  1fk4C  1fk6C  1fk7C  1mzlS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 9062.161 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) | ||||
|---|---|---|---|---|---|
| #2: Chemical | ChemComp-OLA / 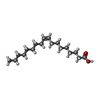  Mass: 282.461 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H34O2 Mass: 282.461 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H34O2 | ||||
| #3: Chemical |   Mass: 46.025 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: CH2O2 Mass: 46.025 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: CH2O2#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 116 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 116 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.34 Å3/Da / Density % sol: 47.46 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop Details: 3.6-4.8M Na formate, VAPOR DIFFUSION, HANGING DROP, temperature 298.0K |
| Crystal grow | *PLUS Temperature: 24.5-25.5 ℃ |
| Components of the solutions | *PLUS Conc.: 3.6-4.8 M / Common name: sodium formate |
-Data collection
| Diffraction | Mean temperature: 287 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Photon Factory  / Beamline: BL-6A / Wavelength: 1 / Beamline: BL-6A / Wavelength: 1 |
| Detector | Type: FUJI / Detector: IMAGE PLATE / Date: Dec 2, 1999 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Highest resolution: 1.3 Å / Num. obs: 18062 / % possible obs: 75.7 % / Redundancy: 5.17 % / Rmerge(I) obs: 0.057 |
| Reflection | *PLUS Num. measured all: 93324 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: molecular replacement Starting model: (PDB CODE:1MZL) Resolution: 1.3→8 Å
| ||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.3→8 Å
| ||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||
| Software | *PLUS Name: SHELXL-97 / Classification: refinement | ||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.3 Å / Lowest resolution: 8 Å | ||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: s_angle_d / Dev ideal: 2.2 |
