 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1fjl: HOMEODOMAIN FROM THE DROSOPHILA PAIRED PROTEIN BOUND TO A DNA OLI... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1fjl | ||||||
|---|---|---|---|---|---|---|---|
| Title | HOMEODOMAIN FROM THE DROSOPHILA PAIRED PROTEIN BOUND TO A DNA OLIGONUCLEOTIDE | ||||||
 Components Components |
| ||||||
 Keywords Keywords | TRANSCRIPTION/DNA / DNA-BINDING PROTEIN / DNA / PAIRED BOX / TRANSCRIPTION REGULATION / TRANSCRIPTION-DNA COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationperiodic partitioning by pair rule gene / HATs acetylate histones / RNA polymerase II transcription regulatory region sequence-specific DNA binding / nervous system development / DNA-binding transcription factor activity, RNA polymerase II-specific / RNA polymerase II cis-regulatory region sequence-specific DNA binding / regulation of transcription by RNA polymerase II / regulation of DNA-templated transcription / positive regulation of transcription by RNA polymerase II / DNA-templated transcription ...periodic partitioning by pair rule gene / HATs acetylate histones / RNA polymerase II transcription regulatory region sequence-specific DNA binding / nervous system development / DNA-binding transcription factor activity, RNA polymerase II-specific / RNA polymerase II cis-regulatory region sequence-specific DNA binding / regulation of transcription by RNA polymerase II / regulation of DNA-templated transcription / positive regulation of transcription by RNA polymerase II / DNA-templated transcription / DNA binding / nucleus Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2 Å X-RAY DIFFRACTION / Resolution: 2 Å | ||||||
 Authors Authors | Wilson, D.S. / Guenther, B. / Desplan, C. / Kuriyan, J. | ||||||
 Citation Citation | Journal: Cell(Cambridge,Mass.) / Year: 1995 Title: High resolution crystal structure of a paired (Pax) class cooperative homeodomain dimer on DNA. Authors: Wilson, D.S. / Guenther, B. / Desplan, C. / Kuriyan, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1fjl.cif.gz | 86.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1fjl.ent.gz | 61.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1fjl.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/fj/1fjlftp://data.pdbj.org/pub/pdb/validation_reports/fj/1fjl | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj






































- Assembly
Assembly
| Deposited unit | 
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 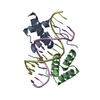
| ||||||||||
| 2 | 
| ||||||||||
| Unit cell |
| ||||||||||
| Details | A COMPLEX OF TWO HOMEODOMAINS (THE PROTEIN COMPONENT) BOUND TO ONE 14 BASE PAIR DNA DUPLEX IS THE MOLECULAR SPECIES ANALYZED IN THIS STUDY. EACH OF THE DNA DUPLEX AND THE COMPLEX HAS PSEUDO TWO-FOLD SYMMETRY ABOUT ITS CENTER. THE ASYMMETRIC UNIT CONTAINS 1.5 COMPLEXES, ONE OF WHICH IS COMPLETELY CONTAINED WITHIN THE ASYMMETRIC UNIT AND THE OTHER OF IS BISECTED AT ITS CENTER OF SYMMETRY BY A CRYSTALLOGRAPHIC TWO-FOLD AXIS. THE TWO-FOLD AVERAGED COMPLEX WAS MODELED AS A COMPLETE SINGLE HOMEODOMAIN AND A COMPLETE SINGLE STRAND OF THE DUPLEX. THEREFORE, THE MODEL CONTAINS THREE HOMEODOMAINS AND THREE DNA STRANDS. |
-Components
| #1: Protein | Mass: 9716.782 Da / Num. of mol.: 3 / Fragment: HOMEODOMAIN / Mutation: C, C4S, S50Q Source method: isolated from a genetically manipulated source Source: (gene. exp.)  #2: DNA chain | | Mass: 4262.816 Da / Num. of mol.: 1 / Source method: obtained synthetically #3: DNA chain | | Mass: 4293.827 Da / Num. of mol.: 1 / Source method: obtained synthetically #4: DNA chain | | Mass: 4269.801 Da / Num. of mol.: 1 / Source method: obtained synthetically #5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 243 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 243 / Source method: isolated from a natural source / Formula: H2OSequence details | THE STANDARD HOMEODOMAIN NUMBERING SYSTEM (SEE, FOR EXAMPLE, KISSINGER, C.R., LIU, B., MARTIN- ...THE STANDARD HOMEODOMAI | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.42 Å3/Da / Density % sol: 49.09 % | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS Temperature: 4 ℃ / pH: 5.5 / Method: unknown | |||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| Reflection | *PLUS Highest resolution: 2 Å / Lowest resolution: 20 Å / Num. obs: 28315 / % possible obs: 92.9 % / Num. measured all: 94960 / Rmerge(I) obs: 0.058 |
| Reflection shell | *PLUS % possible obs: 83.3 % / Rmerge(I) obs: 0.185 |
- Processing
Processing
| Software | Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Rfactor Rwork: 0.198 / Rfactor obs: 0.198 / Highest resolution: 2 Å Details: THE FOLLOWING SIDE CHAINS HAVE BEEN OMITTED FROM THE MODEL BECAUSE THEIR CONFORMATION WAS NOT EVIDENT FROM ELECTRON DENSITY MAPS: HOMEODOMAIN NO. 1: RESIDUES 122, 133, 158. HOMEODOMAIN NO. 2: ...Details: THE FOLLOWING SIDE CHAINS HAVE BEEN OMITTED FROM THE MODEL BECAUSE THEIR CONFORMATION WAS NOT EVIDENT FROM ELECTRON DENSITY MAPS: HOMEODOMAIN NO. 1: RESIDUES 122, 133, 158. HOMEODOMAIN NO. 2: RESIDUES 233, 236, 239, 255, 258. HOMEODOMAIN NO. 3: RESIDUES 300, 324, 357, 358. DUE TO THE TWO-FOLD AVERAGING OF THE DNA DUPLEXES, ALTERNATE SIDE CHAIN IDENTITIES HAVE BEEN MODELED IN THE FOLLOWING WAY: ALTERNATE SIDE CHAIN FOR RESIDUE A D 1A: RESIDUE T D 1B ALTERNATE SIDE CHAIN FOR RESIDUE T D 8A: RESIDUE A D 8B ALTERNATE SIDE CHAIN FOR RESIDUE T E 1A: RESIDUE A E 1B ALTERNATE SIDE CHAIN FOR RESIDUE A E 8A: RESIDUE T E 8B ALTERNATE SIDE CHAIN FOR RESIDUE T F 1A: RESIDUE A F 1B ALTERNATE SIDE CHAIN FOR RESIDUE T F 8A: RESIDUE A F 8B TWO OF THE WATER MOLECULES ARE EXCLUDED BY ONE OF THE TWO ALTERNATE SIDE CHAINS AT DNA RESIDUES NO. 408 AND 508, AS FOLLOWS: WATER RESIDUE NO. 941 IS PRESENT WITH THE ALTERNATE SIDE CHAIN REPRESENTED BY RESIDUE 508 BUT NOT 528. WATER RESIDUE NO. 942 IS PRESENT WITH THE ALTERNATE SIDE CHAIN REPRESENTED BY RESIDUE 428 BUT NOT 408. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Highest resolution: 2 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 6 Å / Rfactor obs: 0.197 / Rfactor Rfree: 0.27 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 38 Å2 |
