 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1fhz | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | PSORALEN CROSS-LINKED D(CCGGTACCGG) FORMS HOLLIDAY JUNCTION | ||||||||||||||||||
 Components Components | DNA (5'-D(* Keywords KeywordsDNA / psoralen / cross-linked DNA / Holliday junction / DNA-drug complex | Function / homology | 4'-HYDROXYMETHYL-4,5',8-TRIMETHYLPSORALEN / DNA |  Function and homology information Function and homology informationMethod |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.2 Å  Authors AuthorsEichman, B.F. / Mooers, B.H.M. / Alberti, M. / Hearst, J.E. / Ho, P.S. |  CitationJournal: J.Mol.Biol. / Year: 2001 CitationJournal: J.Mol.Biol. / Year: 2001Title: The crystal structures of psoralen cross-linked DNAs: drug-dependent formation of Holliday junctions. Authors: Eichman, B.F. / Mooers, B.H. / Alberti, M. / Hearst, J.E. / Ho, P.S. #1: Journal: Proc.Natl.Acad.Sci.USA / Year: 2000Title: The Holliday junction in an inverted repeat DNA sequence: Sequence effects on the structure of four-way junctions Authors: Eichman, B.F. / Vargason, J.M. / Mooers, B.H.M. / Ho, P.S. History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1fhz.cif.gz | 21.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1fhz.ent.gz | 14.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1fhz.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/fh/1fhzftp://data.pdbj.org/pub/pdb/validation_reports/fh/1fhz | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1fhyC C: citing same article ( |
|---|---|
| Similar structure data | |
| Other databases |
|




-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 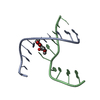
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||
| Unit cell |
| ||||||||||
| Components on special symmetry positions |
| ||||||||||
| Details | A four-way junction is a dimer constructed from chains A and B and a symmetry partner generated by the two-fold |
-Components
| #1: DNA chain | Mass: 3045.992 Da / Num. of mol.: 2 / Source method: obtained synthetically / Details: DNA IS CROSS-LINKED BY PSORALEN #2: Chemical | ChemComp-PSO / | 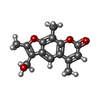  Mass: 258.269 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H14O4 Mass: 258.269 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H14O4#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 43 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 43 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.23 Å3/Da / Density % sol: 44.89 % | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sitting drop / pH: 7 Details: sodium cacodylate, MgCl2, MPD, pH 7, VAPOR DIFFUSION, SITTING DROP, temperature 277K | ||||||||||||||||||||||||||||||||||||
| Components of the solutions |
| ||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion | ||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS  / Beamline: 5.0.2 / Wavelength: 1.1 / Beamline: 5.0.2 / Wavelength: 1.1 |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Oct 8, 1999 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.2→33.39 Å / Num. all: 2900 / Num. obs: 2900 / % possible obs: 96.7 % / Observed criterion σ(I): -3 / Redundancy: 3.3 % / Rmerge(I) obs: 0.067 / Net I/σ(I): 12.3 |
| Reflection shell | Resolution: 2.2→2.28 Å / Redundancy: 3.3 % / Rmerge(I) obs: 0.1 / Num. unique all: 284 / % possible all: 98.3 |
| Reflection | *PLUS Lowest resolution: 33.4 Å / Num. measured all: 9596 |
| Reflection shell | *PLUS % possible obs: 98.3 % / Mean I/σ(I) obs: 8.1 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.2→33.39 Å / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Parkinson et al. Details: ANISOTROPIC B VALUES WERE APPLIED TO FCALC DURING REFINEMENT IN ORDER TO SCALE FCALC TO FOBS
| |||||||||||||||||||||||||
| Displacement parameters |
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.2→33.39 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.851 / Classification: refinement | |||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 33.4 Å / Rfactor Rfree: 0.284 / Rfactor Rwork: 0.222 | |||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
