 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ffq | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF CHITINASE A COMPLEXED WITH ALLOSAMIDIN | |||||||||
 Components Components | CHITINASE A | |||||||||
 Keywords Keywords | HYDROLASE / glycosyl hydrolase / enzyme-inhibitor complex | |||||||||
| Function / homology |  Function and homology information Function and homology informationendochitinase activity / chitinase / chitin catabolic process / chitin binding / polysaccharide catabolic process Similarity search - Function | |||||||||
| Biological species |  Serratia marcescens (bacteria) Serratia marcescens (bacteria) | |||||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.9 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.9 Å | |||||||||
 Authors Authors | Papanikolau, Y. / Tavlas, G. / Vorgias, C.E. / Petratos, K. | |||||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2003 Title: De novo purification scheme and crystallization conditions yield high-resolution structures of chitinase A and its complex with the inhibitor allosamidin. Authors: Papanikolau, Y. / Tavlas, G. / Vorgias, C.E. / Petratos, K. #1: Journal: BIOCHEMISTRY / Year: 2001Title: HIGH RESOLUTION STRUCTURAL ANALYSES OF MUTANT CHITINASE A COMPLEXES WITH SUBSTRATES PROVIDE NEW INSIGHT INTO THE MECHANISM OF CATALYSIS Authors: Papanikolau, Y. / Prag, G. / Tavlas, G. / Vorgias, C.E. / Oppenheim, A.B. / Petratos, K. #2: Journal: Structure / Year: 1994Title: Crystal structure of a bacterial chitinase at 2.3 Angstrom resolution Authors: Perrakis, A. / Tews, I. / Dauter, Z. / Oppenheim, A.B. / Chet, I. / Wilson, K.S. / Vorgias, C.E. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ffq.cif.gz | 135.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ffq.ent.gz | 101.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1ffq.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ff/1ffqftp://data.pdbj.org/pub/pdb/validation_reports/ff/1ffq | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1edqSC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |














-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 58639.590 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Serratia marcescens (bacteria) / Strain: 2170 / Plasmid: PBR322 / Production host: References: GenBank: AB015996, UniProt: P07254*PLUS, chitinase |
|---|---|
| #2: Polysaccharide | 2-acetamido-2-deoxy-beta-D-allopyranose-(1-4)-2-acetamido-2-deoxy-beta-D-allopyranose Source method: isolated from a genetically manipulated source |
| #3: Chemical | ChemComp-AMI / 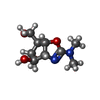  Mass: 216.234 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H16N2O4 Mass: 216.234 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H16N2O4 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 660 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 660 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.3 Å3/Da / Density % sol: 63 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, hanging drop / pH: 7.2 Details: 0.75 M Citrate-Na pH 7.2, 20% (v/v) Methanol, VAPOR DIFFUSION, HANGING DROP, temperature 291K |
-Data collection
| Diffraction | Mean temperature: 110 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU300 / Wavelength: 1.5418 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Sep 14, 1999 / Details: PAIR OF NICKEL MIRRORS |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→10 Å / Num. all: 60767 / Num. obs: 60767 / % possible obs: 98.6 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 3.1 % / Biso Wilson estimate: 24.6 Å2 / Rmerge(I) obs: 0.056 / Net I/σ(I): 22.6 |
| Reflection shell | Resolution: 1.9→1.97 Å / Redundancy: 2.7 % / Rmerge(I) obs: 0.249 / Mean I/σ(I) obs: 4.1 / Num. unique all: 5852 / % possible all: 96.2 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1EDQ Resolution: 1.9→10 Å / SU B: 2.82 / SU ML: 0.08 / σ(F): 0 / σ(I): 0 / ESU R: 0.13 / ESU R Free: 0.13 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 26.3 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|
