 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1f5p: 2.9 ANGSTROM CRYSTAL STRUCTURE OF LAMPREY HEMOGLOBIN THAT HAS BEE... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1f5p | ||||||
|---|---|---|---|---|---|---|---|
| Title | 2.9 ANGSTROM CRYSTAL STRUCTURE OF LAMPREY HEMOGLOBIN THAT HAS BEEN EXPOSED TO CARBON MONOXIDE. | ||||||
 Components Components | HEMOGLOBIN V | ||||||
 Keywords Keywords | OXYGEN STORAGE/TRANSPORT / crystalline ligand transitions / hemoglobin / heme / lamprey / OXYGEN STORAGE-TRANSPORT COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationoxygen carrier activity / oxygen binding / oxidoreductase activity / iron ion binding / heme binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.9 Å X-RAY DIFFRACTION / Resolution: 2.9 Å | ||||||
 Authors Authors | Heaslet, H.A. / Royer Jr., W.E. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2001 Title: Crystalline ligand transitions in lamprey hemoglobin. Structural evidence for the regulation of oxygen affinity. Authors: Heaslet, H.A. / Royer Jr., W.E. #1: Journal: Structure / Year: 1999Title: The 2.7 Angstrom Crystal Structure of Deoxygentated Hemoglobin from the Sea Lamprey (Petromyzon Marinus): Structural Basis for a Lowered Oxygen Affinity and Bohr Effect. Authors: Heaslet, H.A. / Royer Jr., W.E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1f5p.cif.gz | 168.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1f5p.ent.gz | 139.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1f5p.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/f5/1f5pftp://data.pdbj.org/pub/pdb/validation_reports/f5/1f5p | HTTPS FTP |
|---|
-Related structure data













-Links
 PDBj
PDBj











- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | The physiologically relevent assembly is a homodimer in which the subunits are related by a twofold axis of symmetry. / A hexameric assembly is observed upon application of crystallographic symmetry operators. The subunits in the hexamer are arranged as one turn of an approximately three-fold screw. |
-Components
| #1: Protein | Mass: 16289.707 Da / Num. of mol.: 6 / Source method: isolated from a natural source / Source: (natural) #2: Chemical | ChemComp-HEM /   Mass: 616.487 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C34H32FeN4O4 Mass: 616.487 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C34H32FeN4O4#3: Chemical | ChemComp-CMO / 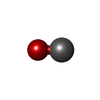  Mass: 28.010 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: CO Mass: 28.010 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: CO#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 6 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 6 / Source method: isolated from a natural source / Formula: H2OCompound details | Crystalline ligand transition experiments. | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.94 Å3/Da / Density % sol: 58.23 % | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 296 K / Method: small tubes / pH: 6.8 Details: 25% PEG 4K, 170mM phosphate buffer, pH 6.8, SMALL TUBES, temperature 296K | |||||||||||||||
| Crystal grow | *PLUS Method: batch method | |||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 296 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS IV / Detector: IMAGE PLATE / Date: Mar 8, 2000 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.9→40 Å / Num. all: 59776 / Num. obs: 19804 / % possible obs: 75.6 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 2 / Redundancy: 3.01 % / Rmerge(I) obs: 0.109 / Net I/σ(I): 7.4 |
| Reflection shell | Resolution: 2.9→3 Å / Redundancy: 9.7 % / Rmerge(I) obs: 0.376 / Num. unique all: 2036 / % possible all: 79.1 |
| Reflection | *PLUS Num. obs: 24593 / % possible obs: 93.9 % / Num. measured all: 81837 / Rmerge(I) obs: 0.118 |
| Reflection shell | *PLUS % possible obs: 79.3 % / Rmerge(I) obs: 0.375 / Mean I/σ(I) obs: 2.2 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.9→10 Å / σ(F): 0 / σ(I): 2 / Stereochemistry target values: Engh & Huber Details: Simulated annealing, Powell minimization, group B-factor and group occupancy refinements using tight non-crystallographic symmetry restraints (wa=200) on the protein except for residue 73.
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.9→10 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.9 / Classification: refinement | |||||||||||||||||||||||||
| Refinement | *PLUS Num. reflection obs: 19647 / Num. reflection Rfree: 2168 / Rfactor Rfree: 0.254 / Rfactor Rwork: 0.206 / Highest resolution: 2.9 Å / Lowest resolution: 8 Å / σ(F): 0 / Rfactor obs: 0.23 | |||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
