 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1ezg: CRYSTAL STRUCTURE OF ANTIFREEZE PROTEIN FROM THE BEETLE, TENEBRIO... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ezg | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF ANTIFREEZE PROTEIN FROM THE BEETLE, TENEBRIO MOLITOR | ||||||
 Components Components | THERMAL HYSTERESIS PROTEIN ISOFORM YL-1 | ||||||
 Keywords Keywords | ANTIFREEZE PROTEIN / insect antifreeze protein / thermal hysteresis / Tenebrio molitor / iodination / right-handed beta-helix / TmAFP | ||||||
| Function / homology | Insect antifreeze protein / Insect antifreeze protein motif / Insect cysteine-rich antifreeze protein / Insect antifreeze protein repeat / Pectate Lyase C-like / 3 Solenoid / extracellular region / Mainly Beta / Thermal hysteresis protein YL-1 Function and homology information Function and homology information | ||||||
| Biological species |  Tenebrio molitor (yellow mealworm) Tenebrio molitor (yellow mealworm) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 1.4 Å X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 1.4 Å | ||||||
 Authors Authors | Liou, Y.-C. / Tocilj, A. / Davies, P.L. / Jia, Z. | ||||||
 Citation Citation | Journal: Nature / Year: 2000 Title: Mimicry of ice structure by surface hydroxyls and water of a beta-helix antifreeze protein. Authors: Liou, Y.C. / Tocilj, A. / Davies, P.L. / Jia, Z. | ||||||
| History |
|
- Structure visualization
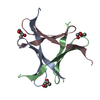
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ezg.cif.gz | 36.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ezg.ent.gz | 26.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1ezg.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ez/1ezgftp://data.pdbj.org/pub/pdb/validation_reports/ez/1ezg | HTTPS FTP |
|---|
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data |






-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | In solution, it is monomer. But it is a dimer in crystal. |
-Components
| #1: Protein | Mass: 8389.097 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Tenebrio molitor (yellow mealworm) / Description: LARVAE HEMOLYMPH / Gene: CDNA FROM FAT BODY LIBRARY / Plasmid: PET20B / Species (production host): Escherichia coli / Production host:  Has protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.49 Å3/Da / Density % sol: 50.62 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / pH: 5.5 Details: ammonium sulfate, sodium citra, cobalt chloridee, pH 5.5, VAPOR DIFFUSION, HANGING DROP, temperature 295K | ||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 9 | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X8C / Wavelength: 0.91 / Beamline: X8C / Wavelength: 0.91 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Apr 10, 1999 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.91 Å / Relative weight: 1 |
| Reflection | Resolution: 1.4→25 Å / Num. all: 133370 / Num. obs: 32557 / % possible obs: 96.5 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 4.5 % / Biso Wilson estimate: 15 Å2 / Rmerge(I) obs: 0.056 / Net I/σ(I): 10.1 |
| Reflection shell | Resolution: 1.4→1.45 Å / Redundancy: 3.7 % / Rmerge(I) obs: 0.208 / Num. unique all: 3086 / % possible all: 95.6 |
| Reflection shell | *PLUS % possible obs: 95.6 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.4→5 Å / σ(F): 0 / σ(I): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.4→5 Å
| ||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||
| Software | *PLUS Name: SHELXL-97 / Classification: refinement | ||||||||||||||||||||
| Refine LS restraints | *PLUS
|
