 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1dy7 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Cytochrome cd1 Nitrite Reductase, CO complex | ||||||
 Components Components | NITRITE REDUCTASE | ||||||
 Keywords Keywords | OXIDOREDUCTASE / ENZYME / NITRITE REDUCTASE | ||||||
| Function / homology |  Function and homology information Function and homology informationhydroxylamine reductase / hydroxylamine reductase activity / nitrite reductase (NO-forming) / nitrite reductase (NO-forming) activity / periplasmic space / electron transfer activity / heme binding / metal ion binding Similarity search - Function | ||||||
| Biological species |  PARACOCCUS PANTOTROPHUS (bacteria) PARACOCCUS PANTOTROPHUS (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.6 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.6 Å | ||||||
 Authors Authors | Sjogren, T. / Svensson-Ek, M. / Hajdu, J. / Brzezinski, P. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2000 Title: Proton-Coupled Structural Changes Upon Binding of Carbon Monoxide to Cytochrome Cd(1): A Combined Flash Photolysis and X-Ray Crystallography Study Authors: Sjogren, T. / Svensson-Ek, M. / Hajdu, J. / Brzezinski, P. #1: Journal: Nature / Year: 1997Title: Haem-Ligand Switching During Catalysis in Crystals of a Nitrogen-Cycle Enzyme Authors: Williams, P.A. / Fulop, V. / Garman, E.F. / Saunders, N.F. / Ferguson, S.J. / Hajdu, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1dy7.cif.gz | 224.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1dy7.ent.gz | 177 KB | Display | PDB format |
| PDBx/mmJSON format | 1dy7.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/dy/1dy7ftp://data.pdbj.org/pub/pdb/validation_reports/dy/1dy7 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1aofS S: Starting model for refinement |
|---|---|
| Similar structure data |














-Links
 PDBj
PDBj













- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 62546.539 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Details: ORGANISM FORMERLY KNOWN AS THIOSPHAERA PANTOTROPHA / Source: (natural) PARACOCCUS PANTOTROPHUS (bacteria) / Cellular location: PERIPLASMReferences: UniProt: P72181, nitrite reductase (NO-forming), hydroxylamine reductase |
|---|
-Non-polymers , 6 types, 858 molecules 
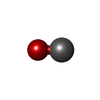




| #2: Chemical |  Mass: 712.484 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C34H32FeN4O10 Mass: 712.484 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C34H32FeN4O10#3: Chemical |  Mass: 28.010 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CO Mass: 28.010 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CO#4: Chemical |  Mass: 92.094 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H8O3#5: Chemical |  Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4#6: Chemical | ChemComp-HEC / |  Mass: 618.503 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H34FeN4O4 Mass: 618.503 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H34FeN4O4#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 847 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has protein modification | Y |
|---|---|
| Sequence details | REFERENCE: THE SEQUENCE IS FROM BAKER S.C., SAUNDERS N.F.W., WILLIS A.C., FERGUSON S.J., FUELOEP V. ...REFERENCE: THE SEQUENCE IS FROM BAKER S.C., SAUNDERS N.F.W., WILLIS A.C., FERGUSON S.J., FUELOEP V., HAJDU J.; J. MOL. BIOL. 269:440-455(1997). THE N-TERMINAL DOMAIN (RESIDUES 1-134) OF CHAIN A IS HIGHLY DISORDERED |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.52 Å3/Da / Density % sol: 51 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / pH: 7 Details: 2.3 M AMMONIUM SULFATE 50MM POTASSIUM PHOSPHATE PH 7.0 CRYSTALS WERE REDUCED USING 20MM SODIUM DITHIONITE. THE CRYSTAL WAS TRANSFERRED TO A SOLUTION CONTAINING 2.3 M AMMONIUM SULFATE, 50 MM ...Details: 2.3 M AMMONIUM SULFATE 50MM POTASSIUM PHOSPHATE PH 7.0 CRYSTALS WERE REDUCED USING 20MM SODIUM DITHIONITE. THE CRYSTAL WAS TRANSFERRED TO A SOLUTION CONTAINING 2.3 M AMMONIUM SULFATE, 50 MM PHOSPHATE BUFFER PH 7 AND 15 % GLYCEROL. CO WAS INTRODUCED UNDER 15 ATM PRESSURE FOR 20 MINUTES AT -20 DEGREES. | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 15 ℃ / Method: vapor diffusion, hanging drop / Details: Fulop, V., (1993) J. Mol. Biol., 232, 1211. | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-3 / Wavelength: 0.935 / Beamline: ID14-3 / Wavelength: 0.935 |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Dec 16, 1998 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.935 Å / Relative weight: 1 |
| Reflection | Resolution: 1.57→30 Å / Num. obs: 148443 / % possible obs: 87.3 % / Observed criterion σ(I): 0 / Redundancy: 2.1 % / Biso Wilson estimate: 15.718 Å2 / Rmerge(I) obs: 0.04 / Rsym value: 0.04 / Net I/σ(I): 12 |
| Reflection shell | Resolution: 1.57→1.64 Å / Rmerge(I) obs: 0.17 / Mean I/σ(I) obs: 2.1 / Rsym value: 0.17 / % possible all: 83.7 |
| Reflection | *PLUS Rmerge(I) obs: 0.04 |
| Reflection shell | *PLUS % possible obs: 83.7 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1AOF Resolution: 1.6→30 Å / SU B: 1.37 / SU ML: 0.049 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.09 / ESU R Free: 0.087 Details: THE N-TERMINAL DOMAIN OF MONOMER A WAS HIGHLY DISORDERED AND WAS NOT MODELLED. IN MONOMER B THE N -TERMINAL RESIDUES B 1 - B 31 ARE DISORDERED IN THE STRUCTURE AND WERE NOT MODELED.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.6→30 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: REFMAC / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.178 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
