 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1duc | ||||||
|---|---|---|---|---|---|---|---|
| Title | EIAV DUTPASE DUDP/STRONTIUM COMPLEX | ||||||
 Components Components | DEOXYURIDINE 5'-TRIPHOSPHATE NUCLEOTIDOHYDROLASE | ||||||
 Keywords Keywords | HYDROLASE / DUTPASE / EIAV / TRIMERIC ENZYME / INHIBITOR COMPLEX / ASPARTYL PROTEASE | ||||||
| Function / homology |  Function and homology information Function and homology informationdUTP catabolic process / dUMP biosynthetic process / dUTP diphosphatase activity / Hydrolases; Acting on peptide bonds (peptidases); Aspartic endopeptidases / UTP binding / retroviral ribonuclease H / exoribonuclease H / exoribonuclease H activity / DNA integration / viral genome integration into host DNA ...dUTP catabolic process / dUMP biosynthetic process / dUTP diphosphatase activity / Hydrolases; Acting on peptide bonds (peptidases); Aspartic endopeptidases / UTP binding / retroviral ribonuclease H / exoribonuclease H / exoribonuclease H activity / DNA integration / viral genome integration into host DNA / RNA-directed DNA polymerase / establishment of integrated proviral latency / telomerase activity / RNA stem-loop binding / RNA-DNA hybrid ribonuclease activity / Transferases; Transferring phosphorus-containing groups; Nucleotidyltransferases / DNA recombination / aspartic-type endopeptidase activity / Hydrolases; Acting on ester bonds / symbiont entry into host cell / magnesium ion binding / proteolysis / DNA binding / zinc ion binding Similarity search - Function | ||||||
| Biological species |  Equine infectious anemia virus Equine infectious anemia virus | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.05 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.05 Å | ||||||
 Authors Authors | Dauter, Z. / Persson, R. / Rosengren, A.M. / Nyman, P.O. / Wilson, K.S. / Cedergren-Zeppezauer, E.S. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 1999 Title: Crystal structure of dUTPase from equine infectious anaemia virus; active site metal binding in a substrate analogue complex. Authors: Dauter, Z. / Persson, R. / Rosengren, A.M. / Nyman, P.O. / Wilson, K.S. / Cedergren-Zeppezauer, E.S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1duc.cif.gz | 41.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1duc.ent.gz | 27.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1duc.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/du/1ducftp://data.pdbj.org/pub/pdb/validation_reports/du/1duc | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1dunC  1dupS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||
| Unit cell |
| |||||||||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 14784.834 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Details: COMPLEX WITH DUDP AND STRONTIUM(II) ION / Source: (gene. exp.) Equine infectious anemia virus / Genus: Lentivirus / Cell line: BL21 / Plasmid: PET-3A/EDU / Production host:  |
|---|---|
| #2: Chemical | ChemComp-SR /   Mass: 87.620 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Sr Mass: 87.620 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Sr |
| #3: Chemical | ChemComp-DUD / 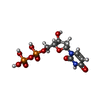  Mass: 388.162 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H14N2O11P2 Mass: 388.162 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H14N2O11P2 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 80 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 80 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.43 Å3/Da / Density % sol: 64 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7 Details: DROP: 3.0 MG/ML PROTEIN, 0.05 M IMIDAZOL MALATE BUFFER, PH 7.0, 21% PEG 400, 20 MM SRCL2, 5 MM DUDP; WELL: 0.1 M IMIDAZOLE MALATE BUFFER, PH 7.0, 42% PEG 400 | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / Method: vapor diffusion, sitting drop | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 277 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: BW7B / Wavelength: 0.885 / Beamline: BW7B / Wavelength: 0.885 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Oct 1, 1995 / Details: MIRRORS |
| Radiation | Monochromator: SI(111) / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.885 Å / Relative weight: 1 |
| Reflection | Resolution: 2.05→25 Å / Num. obs: 13598 / % possible obs: 100 % / Observed criterion σ(I): -3 / Redundancy: 10.5 % / Biso Wilson estimate: 22.9 Å2 / Rmerge(I) obs: 0.124 / Rsym value: 0.124 / Net I/σ(I): 17.7 |
| Reflection shell | Resolution: 2.05→2.09 Å / Redundancy: 10 % / Rmerge(I) obs: 0.686 / Mean I/σ(I) obs: 3.8 / Rsym value: 0.686 / % possible all: 100 |
| Reflection | *PLUS Num. measured all: 142838 |
| Reflection shell | *PLUS % possible obs: 100 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1DUP Resolution: 2.05→20 Å / Cross valid method: FREE R / σ(F): 0 / Details: COORDINATE ERROR ACCORDING TO CRUICKSHANK = 0.11 A
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 27.9 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati d res low obs: 20 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.05→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: REFMAC / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.1686 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
