 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1dlk: CRYSTAL STRUCTURE ANALYSIS OF DELTA-CHYMOTRYPSIN BOUND TO A PEPTI... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1dlk | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE ANALYSIS OF DELTA-CHYMOTRYPSIN BOUND TO A PEPTIDYL CHLOROMETHYL KETONE INHIBITOR | ||||||
 Components Components |
| ||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / Delta-chymotrypsin / peptidic inhibior / chloromethyl ketone / HYDROLASE / HYDROLASE-HYDROLASE INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology informationchymotrypsin / serpin family protein binding / serine protease inhibitor complex / digestion / serine-type endopeptidase activity / proteolysis / extracellular region Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 2.14 Å X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 2.14 Å | ||||||
 Authors Authors | Mac Sweeney, A. / Birrane, G. / Walsh, M.A. / O'Connell, T. / Malthouse, J.P.G. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2000 Title: Crystal structure of delta-chymotrypsin bound to a peptidyl chloromethyl ketone inhibitor. Authors: Mac Sweeney, A. / Birrane, G. / Walsh, M.A. / O'Connell, T. / Malthouse, J.P. / Higgins, T.M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1dlk.cif.gz | 111.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1dlk.ent.gz | 85.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1dlk.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/dl/1dlkftp://data.pdbj.org/pub/pdb/validation_reports/dl/1dlk | HTTPS FTP |
|---|
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (0.03684, -0.49183, 0.86991), Vector: |
-Components
| #1: Protein/peptide | Mass: 1253.511 Da / Num. of mol.: 2 / Fragment: RESIDUES 1-13 / Source method: isolated from a natural source / Source: (natural) #2: Protein | Mass: 24206.271 Da / Num. of mol.: 2 / Fragment: RESIDUES 16-245 / Source method: isolated from a natural source / Source: (natural) #3: Protein/peptide | 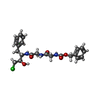  Type: Peptide-like / Class: Enzyme inhibitor / Mass: 466.359 Da / Num. of mol.: 2 / Source method: obtained synthetically / Details: this sequence was synthetically constructed / References: Z-GLY-GLY-PHE-CHLOROMETHYL KETONE (BOUND FORM) Type: Peptide-like / Class: Enzyme inhibitor / Mass: 466.359 Da / Num. of mol.: 2 / Source method: obtained synthetically / Details: this sequence was synthetically constructed / References: Z-GLY-GLY-PHE-CHLOROMETHYL KETONE (BOUND FORM)#4: Chemical | ChemComp-CL / |   Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 281 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 281 / Source method: isolated from a natural source / Formula: H2OCompound details | THE PEPTIDIC INHIBITOR BINDS TO THROMBIN THROUGH TWO COVALENT BONDS: A BOND BETWEEN 0QE AND HIS 57 ...THE PEPTIDIC INHIBITOR BINDS TO THROMBIN THROUGH TWO COVALENT BONDS: A BOND BETWEEN 0QE AND HIS 57 AND A HEMIKETAL BETWEEN HPH AND SER 195. | Has protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.18 Å3/Da / Density % sol: 70.59 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: 2.6 M ammonium sulfate, 200 mM MES-HCl, 10 mM CoCl2, pH 6.5, VAPOR DIFFUSION, HANGING DROP, temperature 298K | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 293 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: X11 / Wavelength: 0.911 / Beamline: X11 / Wavelength: 0.911 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Oct 20, 1996 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.911 Å / Relative weight: 1 |
| Reflection | Resolution: 2.14→40 Å / Num. all: 48065 / Num. obs: 39978 / % possible obs: 82.7 % / Observed criterion σ(I): 2 / Redundancy: 6.6 % / Biso Wilson estimate: 11.2 Å2 / Rmerge(I) obs: 0.081 / Net I/σ(I): 22.6 |
| Reflection shell | Resolution: 2.14→2.18 Å / Redundancy: 5 % / Rmerge(I) obs: 0.43 / Num. unique all: 2370 / % possible all: 100 |
| Reflection | *PLUS Num. obs: 48065 / % possible obs: 99.6 % / Num. measured all: 308959 |
| Reflection shell | *PLUS % possible obs: 100 % / Rmerge(I) obs: 0.435 / Mean I/σ(I) obs: 4.3 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.14→19.9 Å / σ(F): 0 / Stereochemistry target values: John Priestle / Details: Used maximum likelihood procedure
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.14→19.9 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| Software | *PLUS Name: REFMAC / Classification: refinement | |||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 40 Å / Num. reflection obs: 48013 / σ(F): 0 / Num. reflection Rfree: 2370 / % reflection Rfree: 10 % / Rfactor obs: 0.212 / Rfactor Rfree: 0.251 | |||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: p_angle_d / Dev ideal: 2 |
