 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1dcy: CRYSTAL STRUCTURE OF HUMAN S-PLA2 IN COMPLEX WITH INDOLE 3 ACTIVE... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1dcy | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF HUMAN S-PLA2 IN COMPLEX WITH INDOLE 3 ACTIVE SITE INHIBITOR | ||||||
 Components Components | PHOSPHOLIPASE A2 | ||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / S-PLA2 / STRUCTURE-BASED DRUG DESIGN / HYDROLASE-HYDROLASE INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of neutrophil activation / phosphatidylethanolamine metabolic process / phosphatidic acid metabolic process / Acyl chain remodelling of PG / Acyl chain remodelling of PC / Acyl chain remodelling of PI / Acyl chain remodelling of PS / Acyl chain remodelling of PE / Synthesis of PA / intestinal stem cell homeostasis ...regulation of neutrophil activation / phosphatidylethanolamine metabolic process / phosphatidic acid metabolic process / Acyl chain remodelling of PG / Acyl chain remodelling of PC / Acyl chain remodelling of PI / Acyl chain remodelling of PS / Acyl chain remodelling of PE / Synthesis of PA / intestinal stem cell homeostasis / phosphatidylglycerol metabolic process / phosphatidylcholine metabolic process / low-density lipoprotein particle remodeling / phospholipase A2 / positive regulation of macrophage derived foam cell differentiation / A2-type glycerophospholipase activity / Antimicrobial peptides / arachidonate secretion / phospholipid metabolic process / negative regulation of T cell proliferation / lipid catabolic process / secretory granule / angiotensin-activated signaling pathway / phospholipid binding / positive regulation of inflammatory response / killing of cells of another organism / positive regulation of ERK1 and ERK2 cascade / mitochondrial outer membrane / defense response to Gram-positive bacterium / inflammatory response / calcium ion binding / endoplasmic reticulum membrane / perinuclear region of cytoplasm / endoplasmic reticulum / : / extracellular exosome / extracellular region / plasma membrane Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.7 Å X-RAY DIFFRACTION / Resolution: 2.7 Å | ||||||
 Authors Authors | Chirgadze, N.Y. / Schevitz, R.W. / Wery, J.-P. | ||||||
 Citation Citation | Journal: Nat.Struct.Biol. / Year: 1995 Title: Structure-based design of the first potent and selective inhibitor of human non-pancreatic secretory phospholipase A2. Authors: Schevitz, R.W. / Bach, N.J. / Carlson, D.G. / Chirgadze, N.Y. / Clawson, D.K. / D Dillard, R. / Draheim, S.D. / Hartley, L.W. / Jones, N.D. / Mihelich, E.D. / L Olkowski, J. / Snyder, D.W. / ...Authors: Schevitz, R.W. / Bach, N.J. / Carlson, D.G. / Chirgadze, N.Y. / Clawson, D.K. / D Dillard, R. / Draheim, S.D. / Hartley, L.W. / Jones, N.D. / Mihelich, E.D. / L Olkowski, J. / Snyder, D.W. / Dand, S.C. / Wery, J.-P. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1dcy.cif.gz | 37.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1dcy.ent.gz | 25.6 KB | Display | PDB format |
| PDBx/mmJSON format | 1dcy.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/dc/1dcyftp://data.pdbj.org/pub/pdb/validation_reports/dc/1dcy | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 13945.012 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / References: UniProt: P14555, phospholipase A2 | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical |   Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca#3: Chemical | ChemComp-I3N / | 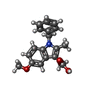  Mass: 309.359 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H19NO3 Mass: 309.359 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H19NO3#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 13 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 13 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.8 Å3/Da / Density % sol: 56 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 297 K / Method: vapor diffusion / pH: 7 Details: 10 MG/ML OF PROTEIN, 50 MM BUFFER (MES OF MOPS), 1% PYRIDINE, AN INHIBITOR CONCENTRATION OF 1.5 MOLAR EQUIVALENT, pH 7.0, VAPOR DIFFUSION, temperature 297K | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS PH range low: 7.5 / PH range high: 6.6 | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 297 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.54 |
| Detector | Type: RIGAKU RAXIS IIC / Detector: IMAGE PLATE / Date: Nov 14, 1994 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.54 Å / Relative weight: 1 |
| Reflection | Resolution: 2.7→30 Å / Num. obs: 4448 / % possible obs: 98.7 % / Redundancy: 5.9 % / Biso Wilson estimate: 25.5 Å2 / Rmerge(I) obs: 0.081 / Net I/σ(I): 27.8 |
| Reflection shell | Resolution: 2.7→2.75 Å / Redundancy: 5.9 % / Rmerge(I) obs: 0.26 / Mean I/σ(I) obs: 6.4 / % possible all: 100 |
| Reflection | *PLUS Highest resolution: 2.2 Å / Lowest resolution: 30 Å / % possible obs: 78 % / Redundancy: 5.9 % / Rmerge(I) obs: 0.092 / Biso Wilson estimate: 25.5 Å2 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.7→20 Å / σ(F): 2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.7→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.7→2.82 Å / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 98 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.2 Å / Rfactor obs: 0.196 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.336 / % reflection Rfree: 3.9 % / Rfactor Rwork: 0.297 |
