 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
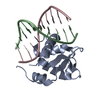
Yorodumi- PDB-1dbu: Crystal structure of cysteinyl-tRNA(Pro) deacylase protein from H... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1dbu | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of cysteinyl-tRNA(Pro) deacylase protein from H. influenzae (HI1434) | ||||||
 Components Components | cysteinyl-tRNA(Pro) deacylase | ||||||
 Keywords Keywords | HYDROLASE / STRUCTURAL GENOMICS / YBAK / Structure 2 Function Project / S2F | ||||||
| Function / homology |  Function and homology information Function and homology informationaminoacyl-tRNA metabolism involved in translational fidelity / Lyases; Carbon-oxygen lyases / Ala-tRNA(Pro) deacylase activity / aminoacyl-tRNA deacylase activity / lyase activity / translation / cytoplasm Similarity search - Function | ||||||
| Biological species |  Haemophilus influenzae (bacteria) Haemophilus influenzae (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 1.8 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 1.8 Å | ||||||
 Authors Authors | Zhang, H. / Huang, K. / Li, Z. / Herzberg, O. / Structure 2 Function Project (S2F) | ||||||
 Citation Citation | Journal: Proteins / Year: 2000 Title: Crystal structure of YbaK protein from Haemophilus influenzae (HI1434) at 1.8 A resolution: functional implications. Authors: Zhang, H. / Huang, K. / Li, Z. / Banerjei, L. / Fisher, K.E. / Grishin, N.V. / Eisenstein, E. / Herzberg, O. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1dbu.cif.gz | 47.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1dbu.ent.gz | 32.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1dbu.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/db/1dbuftp://data.pdbj.org/pub/pdb/validation_reports/db/1dbu | HTTPS FTP |
|---|
-Related structure data









-Links
 PDBj
PDBj













- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 17273.607 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Haemophilus influenzae (bacteria) / Description: IMPACT I SYSTEM FROM NEW ENGLAND BIOLABS / Gene: YbaK / Production host: |
|---|---|
| #2: Chemical | ChemComp-HG /   Mass: 200.590 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Hg Mass: 200.590 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Hg |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 171 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 171 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.99 Å3/Da / Density % sol: 28 % | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS pH: 7 / Method: vapor diffusion, hanging dropDetails: drop consists of equal volume of protein and reservoir solutions | ||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X12C / Wavelength: 1.0091 / Beamline: X12C / Wavelength: 1.0091 |
| Detector | Type: BRANDEIS / Detector: CCD / Date: Oct 27, 1998 |
| Radiation | Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.0091 Å / Relative weight: 1 |
| Reflection | Resolution: 1.8→20 Å / Num. obs: 25089 / % possible obs: 98.7 % / Observed criterion σ(I): -3 / Biso Wilson estimate: 20.9 Å2 / Rmerge(I) obs: 0.059 |
| Reflection shell | Resolution: 1.8→1.86 Å / Rmerge(I) obs: 0.357 / % possible all: 98 |
| Reflection | *PLUS Num. measured all: 47185 |
| Reflection shell | *PLUS % possible obs: 98 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 1.8→20 Å / Cross valid method: free-R / σ(F): 0 / σ(I): 0 / ESU R: 0.173 / ESU R Free: 0.171
| ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→20 Å
| ||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||
| Software | *PLUS Name: REFMAC / Classification: refinement | ||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.282 / Rfactor Rwork: 0.215 |
