 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1cw1: CRYSTAL STRUCTURE OF ISOCITRATE DEHYDROGENASE MUTANT K230M BOUND ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1cw1 | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF ISOCITRATE DEHYDROGENASE MUTANT K230M BOUND TO ISOCITRATE AND MN2+ | ||||||
 Components Components | ISOCITRATE DEHYDROGENASE | ||||||
 Keywords Keywords | OXIDOREDUCTASE / Isocitrate Dehydrogenase / mutant | ||||||
| Function / homology |  Function and homology information Function and homology informationisocitrate dehydrogenase (NADP+) / isocitrate dehydrogenase (NADP+) activity / glyoxylate cycle / guanosine tetraphosphate binding / tricarboxylic acid cycle / electron transport chain / NAD binding / response to oxidative stress / magnesium ion binding / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 2.1 Å X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 2.1 Å | ||||||
 Authors Authors | Stroud, R. / Finer-Moore, J. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2000 Title: Active site water molecules revealed in the 2.1 A resolution structure of a site-directed mutant of isocitrate dehydrogenase. Authors: Cherbavaz, D.B. / Lee, M.E. / Stroud, R.M. / Koshland Jr., D.E. #1: Journal: Biochemistry / Year: 1995Title: Mutational Analysis of the Catalytic Residues Lysine 230 and Tyrosine 160 in the NADP+ Dependent Isocitrate dehydrogenase from Escherichia coli Authors: Lee, M.E. / Dyer, D.H. / Klein, O.D. / Bolduc, J.M. / Stoddard, B.L. #2: Journal: Biochemistry / Year: 1991Title: Catalytic Mechanism of NADP+-dependent Isocitrate Dehydrogenase: Implications from the Structures of Magnesium-isocitrate and NADP+ complexes Authors: Hurley, J.H. / Dean, A.M. / Koshland Jr., D.E. / Stroud, R.M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1cw1.cif.gz | 100.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1cw1.ent.gz | 76.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1cw1.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cw/1cw1ftp://data.pdbj.org/pub/pdb/validation_reports/cw/1cw1 | HTTPS FTP |
|---|
-Related structure data
















-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | The biological assembly is a dimer constructuted from chain A by the crystallographic two-fold operation (Y,X,-Z) |
-Components
| #1: Protein | Mass: 45811.578 Da / Num. of mol.: 1 / Mutation: K230M Source method: isolated from a genetically manipulated source Source: (gene. exp.) References: UniProt: P08200, isocitrate dehydrogenase (NADP+) |
|---|---|
| #2: Chemical | ChemComp-MN /   Mass: 54.938 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mn Mass: 54.938 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mn |
| #3: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 |
| #4: Chemical | ChemComp-ICT / 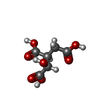  Mass: 192.124 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H8O7 Mass: 192.124 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H8O7 |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 332 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 332 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.33 Å3/Da / Density % sol: 71.57 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 5.8 Details: AMMONIUM SULFATE, DTT, NAN3, MN-ISOCITRATE, pH 5.8, VAPOR DIFFUSION, HANGING DROP, temperature 298.0K | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 110 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL7-1 / Wavelength: 1.08 / Beamline: BL7-1 / Wavelength: 1.08 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.08 Å / Relative weight: 1 |
| Reflection | Resolution: 2.1→5.53 Å / Num. all: 41819 / Num. obs: 40295 / % possible obs: 91.3 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 4.7 % / Biso Wilson estimate: 16.4 Å2 / Rmerge(I) obs: 0.056 / Net I/σ(I): 8.8 |
| Reflection shell | Resolution: 2.1→2.22 Å / % possible all: 89.5 |
| Reflection | *PLUS Num. measured all: 198526 |
| Reflection shell | *PLUS % possible obs: 89.5 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.1→5.53 Å / Rfactor Rfree error: 0.004 / Data cutoff high absF: 13840731.32 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / Stereochemistry target values: ENGH & HUBER
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 17.1 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→5.53 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.1→2.22 Å / Rfactor Rfree error: 0.01 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor Rfree: 0.235 / Rfactor Rwork: 0.189 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Lowest resolution: 2.19 Å / Rfactor Rwork: 0.22 |
