 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1chm: ENZYMATIC MECHANISM OF CREATINE AMIDINOHYDROLASE AS DEDUCED FROM ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1chm | ||||||
|---|---|---|---|---|---|---|---|
| Title | ENZYMATIC MECHANISM OF CREATINE AMIDINOHYDROLASE AS DEDUCED FROM CRYSTAL STRUCTURES | ||||||
 Components Components | CREATINE AMIDINOHYDROLASE | ||||||
 Keywords Keywords | CREATINASE | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  Pseudomonas putida (bacteria) Pseudomonas putida (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 1.9 Å X-RAY DIFFRACTION / Resolution: 1.9 Å | ||||||
 Authors Authors | Hoeffken, H.W. / Knof, S.H. / Bartlett, P.A. / Huber, R. / Moellering, H. / Schumacher, G. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 1990 Title: Enzymatic mechanism of creatine amidinohydrolase as deduced from crystal structures. Authors: Coll, M. / Knof, S.H. / Ohga, Y. / Messerschmidt, A. / Huber, R. / Moellering, H. / Russmann, L. / Schumacher, G. #1: Journal: J.Mol.Biol. / Year: 1988Title: Crystal Structure Determination, Refinement and Molecular Model of Creatine Amidinohydrolase from Pseudomonas Putida Authors: Hoeffken, H.W. / Knof, S.H. / Bartlett, P.A. / Huber, R. / Moellering, H. / Schumacher, G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1chm.cif.gz | 166.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1chm.ent.gz | 131.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1chm.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ch/1chmftp://data.pdbj.org/pub/pdb/validation_reports/ch/1chm | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|









-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Atom site foot note | 1: GLU B 383 - ASN B 384 OMEGA =212.90 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION |
-Components
| #1: Protein | Mass: 45333.125 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Pseudomonas putida (bacteria) / References: UniProt: P38488, creatinase#2: Chemical | 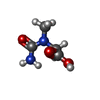  Mass: 132.118 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H8N2O3 Mass: 132.118 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H8N2O3#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 394 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 394 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.27 Å3/Da / Density % sol: 45.8 % | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS pH: 5.4 / Method: batch method | |||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Reflection | *PLUS Highest resolution: 2.37 Å / Lowest resolution: 99 Å / Num. obs: 26026 / Num. measured all: 79941 / Rmerge(I) obs: 0.039 |
|---|---|
| Reflection shell | *PLUS Highest resolution: 2.37 Å / Lowest resolution: 2.52 Å / % possible obs: 41.5 % |
- Processing
Processing
| Software | Name: EREF / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.9→8 Å / Rfactor Rwork: 0.177 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor Rwork: 0.183 / Highest resolution: 2.5 Å / Num. reflection obs: 22809 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
