 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1bwc: STRUCTURE OF HUMAN GLUTATHIONE REDUCTASE COMPLEXED with AJOENE IN... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1bwc | ||||||
|---|---|---|---|---|---|---|---|
| Title | STRUCTURE OF HUMAN GLUTATHIONE REDUCTASE COMPLEXED with AJOENE INHIBITOR AND SUBVERSIVE SUBSTRATE | ||||||
 Components Components | PROTEIN (GLUTATHIONE REDUCTASE) | ||||||
 Keywords Keywords | OXIDOREDUCTASE / FLAVOENZYME / REDOX-ACTIVE CENTER | ||||||
| Function / homology |  Function and homology information Function and homology informationglutathione-disulfide reductase / Metabolism of ingested H2SeO4 and H2SeO3 into H2Se / glutathione-disulfide reductase (NADPH) activity / Interconversion of nucleotide di- and triphosphates / NFE2L2 regulating anti-oxidant/detoxification enzymes / Detoxification of Reactive Oxygen Species / cell redox homeostasis / glutathione metabolic process / TP53 Regulates Metabolic Genes / NADP binding ...glutathione-disulfide reductase / Metabolism of ingested H2SeO4 and H2SeO3 into H2Se / glutathione-disulfide reductase (NADPH) activity / Interconversion of nucleotide di- and triphosphates / NFE2L2 regulating anti-oxidant/detoxification enzymes / Detoxification of Reactive Oxygen Species / cell redox homeostasis / glutathione metabolic process / TP53 Regulates Metabolic Genes / NADP binding / flavin adenine dinucleotide binding / cellular response to oxidative stress / electron transfer activity / mitochondrial matrix / external side of plasma membrane / mitochondrion / extracellular exosome / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.1 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.1 Å | ||||||
 Authors Authors | Gallwitz, H. / Bonse, S. / Martinez-Cruz, A. / Schlichting, I. / Schumacher, K. / Krauth-Siegel, R.L. | ||||||
 Citation Citation | Journal: J.Med.Chem. / Year: 1999 Title: Ajoene is an inhibitor and subversive substrate of human glutathione reductase and Trypanosoma cruzi trypanothione reductase: crystallographic, kinetic, and spectroscopic studies. Authors: Gallwitz, H. / Bonse, S. / Martinez-Cruz, A. / Schlichting, I. / Schumacher, K. / Krauth-Siegel, R.L. #1: Journal: Eur.J.Biochem. / Year: 1988Title: Inhibition of Human Glutathione Reductase by the Nitrosourea Drugs 1,3-Bis(2- Chloroethyl)-1-Nitrosourea and 1-(2-Chloroethyl)-3-(2-Hydroxyethyl)-1- Nitrosourea. A Crystallographic Analysis Authors: Karplus, P.A. / Krauth-Siegel, R.L. / Schirmer, R.H. / Schulz, G.E. #2: Journal: J.Mol.Biol. / Year: 1987Title: Refined Structure of Glutathione Reductase at 1.54 A Resolution Authors: Karplus, P.A. / Schulz, G.E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1bwc.cif.gz | 106.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1bwc.ent.gz | 80.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1bwc.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bw/1bwcftp://data.pdbj.org/pub/pdb/validation_reports/bw/1bwc | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1greS S: Starting model for refinement |
|---|---|
| Similar structure data |












-Links
 PDBj
PDBj









- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 51636.242 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Cell: ERYTHROCYTES / Gene: GOR / Gene (production host): GOR / Production host:  |
|---|---|
| #2: Chemical | ChemComp-CL /   Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl |
| #3: Chemical | ChemComp-FAD /   Mass: 785.550 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C27H33N9O15P2 / Comment: FAD*YM Mass: 785.550 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C27H33N9O15P2 / Comment: FAD*YM |
| #4: Chemical | ChemComp-AJ3 / 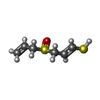  Mass: 162.273 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H10OS2 Mass: 162.273 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H10OS2 |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 142 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 142 / Source method: isolated from a natural source / Formula: H2O |
| Compound details | GLUTATHIONE REDUCTASE IS ACTIVE AS A DIMER OF TWO IDENTICAL SUBUNITS, WHICH ARE COVALENTLY ...GLUTATHION |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.94 Å3/Da / Density % sol: 58.22 % | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6.9 / Details: pH 6.9 | |||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4-25 ℃ / Method: vapor diffusion, hanging drop | |||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 277 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Wavelength: 1.5418 |
| Detector | Type: SIEMENS X1000 / Detector: AREA DETECTOR / Date: Jun 1, 1997 / Details: MONOCHROMATOR |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.1→20 Å / Num. obs: 34707 / % possible obs: 96.4 % / Observed criterion σ(I): 1 / Redundancy: 4.2 % / Biso Wilson estimate: 23.4 Å2 / Rmerge(I) obs: 0.117 |
| Reflection shell | Resolution: 2.1→2.2 Å / % possible all: 74.6 |
| Reflection | *PLUS Lowest resolution: 9999 Å / Num. measured all: 147210 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1GRE Resolution: 2.1→20 Å / Rfactor Rfree error: 0.005 / Data cutoff high absF: 100000 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Details: BULK SOLVENT MODEL USED
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 22.5 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.1→2.2 Å / Rfactor Rfree error: 0.021 / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.851 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS σ(F): 0 / % reflection Rfree: 7.1 % / Rfactor obs: 0.18 / Rfactor Rfree: 0.23 / Rfactor Rwork: 0.18 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 22.5 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.338 / % reflection Rfree: 6.7 % / Rfactor Rwork: 0.285 |
