 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1bsx: STRUCTURE AND SPECIFICITY OF NUCLEAR RECEPTOR-COACTIVATOR INTERACTIONS -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1bsx | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | STRUCTURE AND SPECIFICITY OF NUCLEAR RECEPTOR-COACTIVATOR INTERACTIONS | |||||||||
 Components Components |
| |||||||||
 Keywords Keywords | HORMONE/GROWTH FACTOR / NUCLEAR RECEPTORS / COACTIVATORS / GRIP1 / SPECIFICITY INTERACTION SITE / HORMONE-GROWTH FACTOR COMPLEX | |||||||||
| Function / homology |  Function and homology information Function and homology informationretinal cone cell apoptotic process / female courtship behavior / negative regulation of female receptivity / thyroid hormone binding / retinal cone cell development / thyroid hormone receptor signaling pathway / positive regulation of thyroid hormone receptor signaling pathway / cellular response to thyroid hormone stimulus / regulation of heart contraction / type I pneumocyte differentiation ...retinal cone cell apoptotic process / female courtship behavior / negative regulation of female receptivity / thyroid hormone binding / retinal cone cell development / thyroid hormone receptor signaling pathway / positive regulation of thyroid hormone receptor signaling pathway / cellular response to thyroid hormone stimulus / regulation of heart contraction / type I pneumocyte differentiation / retinoic acid receptor signaling pathway / SUMOylation of intracellular receptors / sensory perception of sound / Nuclear Receptor transcription pathway / chromatin DNA binding / mRNA transcription by RNA polymerase II / nuclear receptor activity / RNA polymerase II transcription regulator complex / transcription coactivator binding / sequence-specific double-stranded DNA binding / DNA-binding transcription factor activity, RNA polymerase II-specific / cell differentiation / nuclear body / RNA polymerase II cis-regulatory region sequence-specific DNA binding / DNA-binding transcription factor activity / chromatin / enzyme binding / negative regulation of transcription by RNA polymerase II / DNA-templated transcription / positive regulation of transcription by RNA polymerase II / DNA binding / zinc ion binding / nucleoplasm / nucleus Similarity search - Function | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 3.7 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 3.7 Å | |||||||||
 Authors Authors | Wagner, R.L. / Darimont, B.D. / Apriletti, J.W. / Stallcup, M.R. / Kushner, P.J. / Baxter, J.D. / Fletterick, R.J. / Yamamoto, K.R. | |||||||||
 Citation Citation | Journal: Genes Dev. / Year: 1998 Title: Structure and specificity of nuclear receptor-coactivator interactions. Authors: Darimont, B.D. / Wagner, R.L. / Apriletti, J.W. / Stallcup, M.R. / Kushner, P.J. / Baxter, J.D. / Fletterick, R.J. / Yamamoto, K.R. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1bsx.cif.gz | 102.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1bsx.ent.gz | 80.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1bsx.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bs/1bsxftp://data.pdbj.org/pub/pdb/validation_reports/bs/1bsx | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (0.52697, 0.84909, -0.03674), Vector: |
-Components
| #1: Protein | Mass: 29410.996 Da / Num. of mol.: 2 / Fragment: LIGAND BINDING DOMAIN Source method: isolated from a genetically manipulated source Details: HIS-TAGGED (THE FOLLOWING RESIDUES WERE NOT SEEN IN THE ELECTRON DENSITY: MGSSHHHHHHSSGLVPRGSHM) Source: (gene. exp.) Homo sapiens (human) / Plasmid: PET28 / Species (production host): Escherichia coli / Production host:  #2: Protein/peptide | Mass: 1507.738 Da / Num. of mol.: 2 / Fragment: NR-BOX2 FROM NUCLEAR RECEPTOR INTERACTION DOMAIN / Source method: obtained synthetically #3: Chemical | 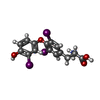  Mass: 650.973 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C15H12I3NO4 / Comment: hormone*YM Mass: 650.973 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C15H12I3NO4 / Comment: hormone*YM |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.91 Å3/Da / Density % sol: 52 % | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 4.9 / Details: pH 4.9 | |||||||||||||||||||||||||||||||||||
| Crystal | *PLUS | |||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / Method: vapor diffusion, hanging drop | |||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 105 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: RIGAKU / Detector: IMAGE PLATE / Date: Nov 15, 1997 |
| Radiation | Monochromator: NI FILTER / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 3.6→25 Å / Num. obs: 8490 / % possible obs: 96.3 % / Observed criterion σ(I): -3 / Redundancy: 4 % / Rmerge(I) obs: 0.077 / Rsym value: 0.077 / Net I/σ(I): 12 |
| Reflection shell | Resolution: 3.6→3.66 Å / Rmerge(I) obs: 0.261 / Mean I/σ(I) obs: 3.7 / Rsym value: 0.261 / % possible all: 96.3 |
| Reflection | *PLUS Num. measured all: 35565 |
| Reflection shell | *PLUS % possible obs: 96.3 % / Num. unique obs: 411 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: HUMAN THYROID HORMONE RECEPTOR BETA Resolution: 3.7→100 Å / Data cutoff high rms absF: 10000 / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 36 Å2 / ksol: 0.27 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.7→100 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | NCS model details: STRICT | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 3.7 Å / Num. reflection all: 7851 / Num. reflection obs: 7614 / σ(F): 2 / % reflection Rfree: 10 % / Rfactor all: 0.254 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
