 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1bq4: SACCHAROMYCES CEREVISIAE PHOSPHOGLYCERATE MUTASE IN COMPLEX WITH ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1bq4 | ||||||
|---|---|---|---|---|---|---|---|
| Title | SACCHAROMYCES CEREVISIAE PHOSPHOGLYCERATE MUTASE IN COMPLEX WITH BENZENE HEXACARBOXYLATE | ||||||
 Components Components | PROTEIN (PHOSPHOGLYCERATE MUTASE 1) | ||||||
 Keywords Keywords | ISOMERASE / TRANSFERASE (PHOSPHORYL) / GLYCOLYTIC ENZYME | ||||||
| Function / homology |  Function and homology information Function and homology informationGluconeogenesis / Glycolysis / phosphoglycerate mutase (2,3-diphosphoglycerate-dependent) / phosphoglycerate mutase activity / canonical glycolysis / Neutrophil degranulation / gluconeogenesis / glycolytic process / mitochondrial intermembrane space / mitochondrial outer membrane ...Gluconeogenesis / Glycolysis / phosphoglycerate mutase (2,3-diphosphoglycerate-dependent) / phosphoglycerate mutase activity / canonical glycolysis / Neutrophil degranulation / gluconeogenesis / glycolytic process / mitochondrial intermembrane space / mitochondrial outer membrane / mitochondrion / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / OTHER / Resolution: 2.5 Å X-RAY DIFFRACTION / OTHER / Resolution: 2.5 Å | ||||||
 Authors Authors | Rigden, D.J. / Phillips, S.E.V. / Fothergill-Gilmore, L.A. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 1999 Title: Polyanionic inhibitors of phosphoglycerate mutase: combined structural and biochemical analysis. Authors: Rigden, D.J. / Walter, R.A. / Phillips, S.E. / Fothergill-Gilmore, L.A. #1: Journal: J.Mol.Biol. / Year: 1999Title: Sulphate Ions Observed in the 2.12 A Structure of a New Crystal Form of S. Cerevisiae Phosphoglycerate Mutase Provide Insights into Understanding the Catalytic Mechanism Authors: Rigden, D.J. / Phillips, S.E.V. / Fothergill-Gilmore, L.A. #2: Journal: J.Mol.Biol. / Year: 1998Title: The 2.3 Angstroms X-Ray Structure of S.Cerevisiae Phosphoglycerate Mutase Authors: Rigden, D.J. / Alexeev, D. / Phillips, S.E.V. / Fothergill-Gilmore, L.A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1bq4.cif.gz | 193.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1bq4.ent.gz | 156.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1bq4.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bq/1bq4ftp://data.pdbj.org/pub/pdb/validation_reports/bq/1bq4 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1bq3C  5pgmS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj











- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (1, 0.00068, -5.0E-5), Vector: |
-Components
| #1: Protein | Mass: 27517.369 Da / Num. of mol.: 4 / Source method: isolated from a natural source / Source: (natural) #2: Chemical |   Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4#3: Chemical | ChemComp-BHC / | 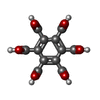  Mass: 342.169 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H6O12 Mass: 342.169 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H6O12Nonpolymer details | HETEROGEN: BHC MODELLED AS SEMI-IONIZED | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.89 Å3/Da / Density % sol: 57.1 % | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 8.65 Details: PROTEIN WAS CRYSTALLISED FROM 22-24% PEG 4000 60MM TRIS-HCL, pH 8.65 | ||||||||||||||||||||||||||||||||||||
| Crystal | *PLUS | ||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, sitting drop / Details: Rigden, D.J., (1998) J.Mol.Biol., 276, 449. | ||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS / Detector: IMAGE PLATE / Date: Jan 15, 1998 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.5→30 Å / Num. obs: 49354 / % possible obs: 63 % / Redundancy: 1.7 % / Rmerge(I) obs: 0.097 / Net I/σ(I): 6 |
| Reflection shell | Resolution: 2.5→2.6 Å / Redundancy: 1.3 % / Rmerge(I) obs: 0.254 / Mean I/σ(I) obs: 2.7 / % possible all: 47.5 |
| Reflection | *PLUS % possible obs: 63 % |
| Reflection shell | *PLUS % possible obs: 47.5 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: OTHER Starting model: PDB ENTRY 5PGM Resolution: 2.5→30 Å / Rfactor Rfree error: 0.0051 / Data cutoff high absF: 100000 / Data cutoff low absF: 0.001 / Cross valid method: THROUGHOUT / σ(F): 0 Details: C-TERMINAL RESIDUES A 235 - A 246, B 236 - B 246, C 236 - C 246 AND D 235 - D 246 ARE NOT VISIBLE IN ELECTRON DENSITY MAP. CLEAVAGE OF SOME OR ALL OF THESE RESIDUES MAY HAVE OCCURRED.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 29.8 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati d res low obs: 30 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.5→30 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | NCS model details: CONSTRAINTS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.5→2.61 Å / Rfactor Rfree error: 0.031 / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.851 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.5 Å / Lowest resolution: 30 Å / σ(F): 0 / % reflection Rfree: 5 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 29.8 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Highest resolution: 2.5 Å / Rfactor Rfree: 0.419 / % reflection Rfree: 5 % / Rfactor Rwork: 0.33 |
