 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1boz: STRUCTURE-BASED DESIGN AND SYNTHESIS OF LIPOPHILIC 2,4-DIAMINO-6-... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1boz | ||||||
|---|---|---|---|---|---|---|---|
| Title | STRUCTURE-BASED DESIGN AND SYNTHESIS OF LIPOPHILIC 2,4-DIAMINO-6-SUBSTITUTED QUINAZOLINES AND THEIR EVALUATION AS INHIBITORS OF DIHYDROFOLATE REDUCTASE AND POTENTIAL ANTITUMOR AGENTS | ||||||
 Components Components | PROTEIN (DIHYDROFOLATE REDUCTASE) | ||||||
 Keywords Keywords | OXIDOREDUCTASE | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of removal of superoxide radicals / tetrahydrobiopterin biosynthetic process / Metabolism of folate and pterines / tetrahydrofolate metabolic process / response to methotrexate / sequence-specific mRNA binding / folic acid binding / axon regeneration / dihydrofolate metabolic process / dihydrofolate reductase ...regulation of removal of superoxide radicals / tetrahydrobiopterin biosynthetic process / Metabolism of folate and pterines / tetrahydrofolate metabolic process / response to methotrexate / sequence-specific mRNA binding / folic acid binding / axon regeneration / dihydrofolate metabolic process / dihydrofolate reductase / G1/S-Specific Transcription / dihydrofolate reductase activity / folic acid metabolic process / tetrahydrofolate biosynthetic process / NADPH binding / 'de novo' pyrimidine nucleobase biosynthetic process / one-carbon metabolic process / Tetrahydrobiopterin (BH4) synthesis, recycling, salvage and regulation / mRNA regulatory element binding translation repressor activity / replication fork / NADP binding / DNA replication / negative regulation of translation / mRNA binding / mitochondrion / nucleus / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.1 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.1 Å | ||||||
 Authors Authors | Gangjee, A. / Vidwans, A.P. / Vasudevan, A. / Queener, S.F. / Kisliuk, R.L. / Cody, V. / Li, R. / Galitsky, N. / Luft, J.R. / Pangborn, W. | ||||||
 Citation Citation | Journal: J.Med.Chem. / Year: 1998 Title: Structure-based design and synthesis of lipophilic 2,4-diamino-6-substituted quinazolines and their evaluation as inhibitors of dihydrofolate reductases and potential antitumor agents. Authors: Gangjee, A. / Vidwans, A.P. / Vasudevan, A. / Queener, S.F. / Kisliuk, R.L. / Cody, V. / Li, R. / Galitsky, N. / Luft, J.R. / Pangborn, W. #1: Journal: Acta Crystallogr.,Sect.D / Year: 1997Title: Comparison of Ternary Complexes of Pneumocystis carinii and Wild-Type Human Dihydrofolate Reductase With a Novel Classical Antitumor Furo[2,3-d]pyrimidine Antifolate Authors: Cody, V. / Galitsky, N. / Luft, J.R. / Pangborn, W. / Gangjee, A. / Devraj, R. / Queener, S.F. / Blakley, R.L. #2: Journal: Biochemistry / Year: 1997Title: Comparison of Two Independent Crystal Structures of Human Dihydrofolate Reductase Ternary Complexes Reduced with Nicotinamide Adenine Dinucleotide Phosphate and the Very Tight-Binding Inhibitor Pt523 Authors: Cody, V. / Galitsky, N. / Luft, J.R. / Pangborn, W. / Rosowsky, A. / Blakley, R.L. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1boz.cif.gz | 56 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1boz.ent.gz | 40 KB | Display | PDB format |
| PDBx/mmJSON format | 1boz.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bo/1bozftp://data.pdbj.org/pub/pdb/validation_reports/bo/1boz | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 21259.402 Da / Num. of mol.: 1 / Mutation: F31G / Source method: isolated from a natural source / Source: (natural) Homo sapiens (human) / References: UniProt: P00374, dihydrofolate reductase |
|---|---|
| #2: Chemical | ChemComp-NDP / 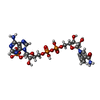  Mass: 745.421 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H30N7O17P3 Mass: 745.421 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H30N7O17P3 |
| #3: Chemical | ChemComp-PRD / 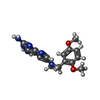  Mass: 340.380 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H20N6O2 Mass: 340.380 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H20N6O2 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 125 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 125 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.61 Å3/Da / Density % sol: 52.93 % | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6.5 / Details: pH 6.5 | |||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / pH: 7 / Method: vapor diffusion, hanging drop | |||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 287 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS / Detector: IMAGE PLATE |
| Radiation | Monochromator: NI FILTER / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.1→53 Å / Num. obs: 9706 / % possible obs: 95 % / Observed criterion σ(I): 2 / Redundancy: 2.5 % / Rmerge(I) obs: 0.049 |
| Reflection shell | Highest resolution: 2.1 Å / Redundancy: 2.5 % / Rmerge(I) obs: 0.125 / % possible all: 78 |
| Reflection | *PLUS % possible obs: 86.4 % / Rmerge(I) obs: 0.0496 |
| Reflection shell | *PLUS Highest resolution: 2 Å / Lowest resolution: 2.1 Å / % possible obs: 63.6 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.1→8 Å / Cross valid method: THROUGHOUT / σ(F): 2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 19.34 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: PROLSQ / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.1 Å / σ(F): 2 / Rfactor obs: 0.202 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
