 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1bnk | ||||||
|---|---|---|---|---|---|---|---|
| Title | HUMAN 3-METHYLADENINE DNA GLYCOSYLASE COMPLEXED TO DNA | ||||||
 Components Components |
| ||||||
 Keywords Keywords | HYDROLASE/DNA / DNA REPAIR / DNA GLYCOSYLASE / HYDROLASE-DNA COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationalkylbase DNA N-glycosylase activity / DNA-3-methyladenine glycosylase II / depurination / DNA N-glycosylase activity / Displacement of DNA glycosylase by APEX1 / DNA alkylation repair / mitochondrial nucleoid / Recognition and association of DNA glycosylase with site containing an affected purine / Cleavage of the damaged purine / base-excision repair ...alkylbase DNA N-glycosylase activity / DNA-3-methyladenine glycosylase II / depurination / DNA N-glycosylase activity / Displacement of DNA glycosylase by APEX1 / DNA alkylation repair / mitochondrial nucleoid / Recognition and association of DNA glycosylase with site containing an affected purine / Cleavage of the damaged purine / base-excision repair / damaged DNA binding / mitochondrion / DNA binding / nucleoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.7 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.7 Å | ||||||
 Authors Authors | Lau, A.Y. / Schaerer, O.D. / Samson, L. / Verdine, G.L. / Ellenberger, T. | ||||||
 Citation Citation | Journal: Cell(Cambridge,Mass.) / Year: 1998 Title: Crystal structure of a human alkylbase-DNA repair enzyme complexed to DNA: mechanisms for nucleotide flipping and base excision. Authors: Lau, A.Y. / Scharer, O.D. / Samson, L. / Verdine, G.L. / Ellenberger, T. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1bnk.cif.gz | 67.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1bnk.ent.gz | 46.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1bnk.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bn/1bnkftp://data.pdbj.org/pub/pdb/validation_reports/bn/1bnk | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj






































- Assembly
Assembly
| Deposited unit | 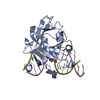
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: DNA chain | Mass: 3832.501 Da / Num. of mol.: 1 / Source method: obtained synthetically |
|---|---|
| #2: DNA chain | Mass: 3975.611 Da / Num. of mol.: 1 / Source method: obtained synthetically |
| #3: Protein | Mass: 24051.623 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Organ: LIVER / Plasmid: PLM1-AAG / Species (production host): Escherichia coli / Cellular location (production host): CYTOPLASM / Gene (production host): AAG / Production host:  References: UniProt: P29372, DNA-3-methyladenine glycosylase II |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 41 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 41 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.49 Å3/Da / Density % sol: 56 % Description: DATA WERE COLLECTED USING THE OSCILLATION METHOD | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7.5 / Details: pH 7.5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 22 ℃ / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 113 K | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X12C / Wavelength: 1.5418, 1.0100, 0.9787, 0.9784, 0.9500 / Beamline: X12C / Wavelength: 1.5418, 1.0100, 0.9787, 0.9784, 0.9500 | ||||||||||||||||||
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Mar 15, 1998 / Details: MIRRORS | ||||||||||||||||||
| Radiation | Monochromator: NI FILTER / Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||
| Radiation wavelength |
| ||||||||||||||||||
| Reflection | Resolution: 2.7→30 Å / Num. obs: 9321 / % possible obs: 100 % / Redundancy: 9 % / Rsym value: 0.046 / Net I/σ(I): 15 | ||||||||||||||||||
| Reflection shell | Resolution: 2.7→2.82 Å / Redundancy: 9 % / Mean I/σ(I) obs: 10 / Rsym value: 0.145 / % possible all: 100 | ||||||||||||||||||
| Reflection | *PLUS % possible obs: 99.6 % / Num. measured all: 269241 / Rmerge(I) obs: 0.046 | ||||||||||||||||||
| Reflection shell | *PLUS % possible obs: 100 % / Rmerge(I) obs: 0.176 / Mean I/σ(I) obs: 12.2 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 2.7→8 Å / Data cutoff high rms absF: 10000 / Cross valid method: THROUGHOUT / σ(F): 3
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.7→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.7→2.79 Å / Total num. of bins used: 10 /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.3C / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.7 Å / Lowest resolution: 8 Å / σ(F): 3 / % reflection Rfree: 10 % / Rfactor Rfree: 0.26 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.302 / Rfactor Rwork: 0.265 |
