 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1af0 | ||||||
|---|---|---|---|---|---|---|---|
| Title | SERRATIA PROTEASE IN COMPLEX WITH INHIBITOR | ||||||
 Components Components | SERRATIA PROTEASE | ||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / METALLOPROTEASE / SERRALYSIN / HYDROLASE-HYDROLASE INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology informationserralysin / symbiont-mediated killing of host cell / metalloendopeptidase activity / toxin activity / extracellular matrix / calcium ion binding / proteolysis / : / extracellular region / zinc ion binding Similarity search - Function | ||||||
| Biological species |  Serratia marcescens (bacteria) Serratia marcescens (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / DIFFERENCE FOURIER / Resolution: 1.8 Å X-RAY DIFFRACTION / DIFFERENCE FOURIER / Resolution: 1.8 Å | ||||||
 Authors Authors | Baumann, U. | ||||||
 Citation Citation | Journal: To be Published Title: Crystal Structure of the 50 kDa Metallo Protease from S. Marcescens in Complex with the Synthetic Inhibitor Cbz-Leu-Ala-Nhoh Authors: Baumann, U. #1: Journal: J.Mol.Biol. / Year: 1995Title: Crystal Structure of a Complex between Serratia Marcescens Metallo-Protease and an Inhibitor from Erwinia Chrysanthemi Authors: Baumann, U. / Bauer, M. / Letoffe, S. / Delepelaire, P. / Wandersman, C. #2: Journal: J.Mol.Biol. / Year: 1994Title: Crystal Structure of the 50 kDa Metallo Protease from Serratia Marcescens Authors: Baumann, U. #3: Journal: Embo J. / Year: 1993Title: Three-Dimensional Structure of the Alkaline Protease of Pseudomonas Aeruginosa: A Two-Domain Protein with a Calcium Binding Parallel Beta Roll Motif Authors: Baumann, U. / Wu, S. / Flaherty, K.M. / Mckay, D.B. #4: Journal: Biochemistry / Year: 1997 Title: Kinetic characterization of the serralysins: a divergent catalytic mechanism pertaining to astacin-type metalloproteases. Authors: Mock, W.L. / Yao, J. | ||||||
| History |
| ||||||
| Remark 650 | HELIX DETERMINATION METHOD: TAKEN FROM PDB ENTRY 1SAT | ||||||
| Remark 700 | SHEET DETERMINATION METHOD: TAKEN FROM PDB ENTRY 1SAT |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1af0.cif.gz | 157.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1af0.ent.gz | 117 KB | Display | PDB format |
| PDBx/mmJSON format | 1af0.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/af/1af0ftp://data.pdbj.org/pub/pdb/validation_reports/af/1af0 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1satS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 50413.156 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Details: PURCHASED FROM SIGMA / Source: (natural) Serratia marcescens (bacteria) / Strain: NOT KNOWN, PROBABLY SM6 / References: UniProt: P23694, serralysin | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical | ChemComp-ZN /   Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn | ||||||
| #3: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: Ca#4: Chemical | ChemComp-0Z9 / | 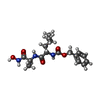  Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 351.398 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H25N3O5 / References: CBZ-LEU-ALA-NHOH Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 351.398 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H25N3O5 / References: CBZ-LEU-ALA-NHOH#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 206 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 206 / Source method: isolated from a natural source / Formula: H2ONonpolymer details | THERE IS A DISORDERED N-CARBO-BENZYLOXY GROUP IN THIS STRUCTURE THAT IS ATTACHED TO LEU B 674. NO ...THERE IS A DISORDERED | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.53 Å3/Da / Density % sol: 62 % |
|---|---|
| Crystal grow | pH: 6.5 / Details: 0.2 M AMMONIUM SULFATE, 20%PEG 4K, PH 6.5 |
-Data collection
| Diffraction | Mean temperature: 290 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH2R / Wavelength: 1.5418 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Oct 1, 1995 / Details: MIRRORS |
| Radiation | Monochromator: NI FILTER / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.8→25 Å / Num. obs: 61944 / % possible obs: 93.7 % / Observed criterion σ(I): 0 / Redundancy: 3.5 % / Rmerge(I) obs: 0.058 / Rsym value: 0.058 / Net I/σ(I): 15.6 |
| Reflection shell | Resolution: 1.8→1.85 Å / Redundancy: 2.4 % / Rmerge(I) obs: 0.251 / Mean I/σ(I) obs: 3.4 / Rsym value: 0.251 / % possible all: 81.7 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: DIFFERENCE FOURIER Starting model: PDB ENTRY 1SAT Resolution: 1.8→30 Å / Num. parameters: 15163 / Num. restraintsaints: 14700 / Cross valid method: FREE R / σ(F): 0 / Stereochemistry target values: ENGH AND HUBER Details: ZN++ AND CA++ IONS REFINED ANISOTROPICALLY. THE N-CARBOBENZYLOXY GROUP OF THE INHIBITOR IS INVISIBLE.
| |||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: MOEWS & KRETSINGER | |||||||||||||||||||||||||||||||||
| Refine analyze | Occupancy sum non hydrogen: 3780 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→30 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
|
